第三节 甾体激素
甾体激素的定义:甾体激素是一类四环脂烃化合物,具有环戊烷多氢菲母核。
甾体激素的分类:按其药理作用,可分为性激素及皮质激素;
根据其化学结构,则可分为雌甾烷、雄甾烷及孕甾烷三大类,其化学结构如下:
甾体激素的分类:按其药理作用,可分为性激素及皮质激素;
根据其化学结构,则可分为雌甾烷、雄甾烷及孕甾烷三大类,其化学结构如下:

结构特点:它们均具有环戊烷多氢菲母核。其中雌甾烷在C-13上有甲基取代,此甲基编号为C-18;雄甾烷及孕甾烷在C-10及C-13上有甲基取代,分别为C-18、C-19;孕甾烷在C-17上有两个碳取代,分别为C-20、C-21。四元环的A、B、C、D还可按以下构型分布:
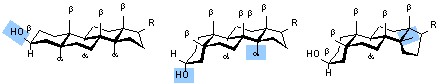
即反-反-反、顺-反-反及顺-反-顺式,此时在环平面以上的取代基(平面式中的实线)称β位取代,在环平面以下的取代基(平面式中的虚线)称α位取代。本节中介绍的甾体激素药物都是反-反-反式构型的化合物。
发展过程:1932~1939年间,从腺体中获得雄酮(estrone 1932年)、雌二醇(estradiol 1932年)、睾酮(testosterone 1935年)及皮质酮(corticosterone 1939年)等的纯品结晶,之后阐明了其化学结构,从此开创了甾体化学和甾体药物化学的新领域。随后,又有许多重大的成就,发明用薯芋皂甙(diosgenin)为原料进行半合成生产甾体药物,使生产规模扩大,成本降低。发现肾上腺皮质激素治疗风湿性关节炎及其在免疫调节上的重要价值,使甾体药物成为医院中不可缺少的药物。甾体口服避孕药的研究成功,使人类生育控制达到了新水平。其中18-甲基炔诺孕酮用全合成方法制得,从而摆脱了完全依靠天然来源的状况,开创了甾体全合成的新局面。用生物合成法在甾体中引入11-氧,以及皮质激素构效关系的研究,都充实了药物化学的基础及应用内容。
发展过程:1932~1939年间,从腺体中获得雄酮(estrone 1932年)、雌二醇(estradiol 1932年)、睾酮(testosterone 1935年)及皮质酮(corticosterone 1939年)等的纯品结晶,之后阐明了其化学结构,从此开创了甾体化学和甾体药物化学的新领域。随后,又有许多重大的成就,发明用薯芋皂甙(diosgenin)为原料进行半合成生产甾体药物,使生产规模扩大,成本降低。发现肾上腺皮质激素治疗风湿性关节炎及其在免疫调节上的重要价值,使甾体药物成为医院中不可缺少的药物。甾体口服避孕药的研究成功,使人类生育控制达到了新水平。其中18-甲基炔诺孕酮用全合成方法制得,从而摆脱了完全依靠天然来源的状况,开创了甾体全合成的新局面。用生物合成法在甾体中引入11-氧,以及皮质激素构效关系的研究,都充实了药物化学的基础及应用内容。
一、甾体雌激素
天然雌激素有雌二醇(estradiol)、雌酮(estrone)及雌三醇(estriol)。其结构特征都是A环芳香类甾体化合物,其生理作用为促进雌性动物第二性征的发育和性器官的成熟,还与孕激素一起完成性周期、妊娠、哺乳等。临床上用于治疗女性性功能疾病、更年期综合征、骨质疏松症,作为口服避孕药及对预防放射线、脂质的代谢都有十分重要的作用。
雌激素可分类为甾体雌激素及非甾体雌激素两大类。
雌激素可分类为甾体雌激素及非甾体雌激素两大类。
代表药物 雌二醇 estradiol
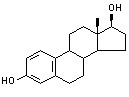
化学名:雄甾-1,3,5(10)-三烯-3,17β-二醇((17β)-estra-1,3,5(10)-triene-3,17-diol)。
体内代谢:在体内,estradiol经代谢羟化或氧化后,与葡萄糖醛酸或硫酸酯结合成为水溶性化合物从尿中排出。
结构改造:estradiol有极强的生物活性,11-8~11-10mol/L的浓度对靶器官即能表现出活性。因而,以estradiol为先导物的结构改造的主要目的往往不是为了提高活性,而是为了使用方便,如能够口服,或长效,或其他的专一用途。
炔雌醇(ethinylestradiol)是一种口服有效的化合物,其口服活性是estradiol的10~20倍,这可能是由于17α位引入乙炔基之后,在肝中17β羟基的硫酸酯化代谢受阻,在胃肠道中也可抵御微生物的降解作用所致。现已成为口服甾体避孕药中最常用的雌激素组分。
体内代谢:在体内,estradiol经代谢羟化或氧化后,与葡萄糖醛酸或硫酸酯结合成为水溶性化合物从尿中排出。
结构改造:estradiol有极强的生物活性,11-8~11-10mol/L的浓度对靶器官即能表现出活性。因而,以estradiol为先导物的结构改造的主要目的往往不是为了提高活性,而是为了使用方便,如能够口服,或长效,或其他的专一用途。
炔雌醇(ethinylestradiol)是一种口服有效的化合物,其口服活性是estradiol的10~20倍,这可能是由于17α位引入乙炔基之后,在肝中17β羟基的硫酸酯化代谢受阻,在胃肠道中也可抵御微生物的降解作用所致。现已成为口服甾体避孕药中最常用的雌激素组分。

进一步将它的3-羟基醚化如甲醚化,特别是环戊醚化后的产物炔雌醇-3-环戊醚(炔雌醚,quinestrol),不但保持了口服活性,醚化产物的脂溶性增大,能在体内脂肪小球中贮存,慢慢降解后离解出3-羟基化合物而起作用,由于醚键在体内的代谢更加复杂及缓慢,因而它是一种口服及注射长效雌激素。我国开发的一种长效口服雌激素尼尔雌醇(nilestriol),它是乙炔雌三醇的环戊醚,雌激素活性小于炔雌醇。口服一片5mg可延效一个月。药物进入体内后缓慢地进行脱烷基化,生成3-羟基化合物后发挥作用。
estradiol的3或17β位羟基的各种各样的酯化产物是最常见的衍生物,它们能在植物油中溶解制成长效针剂。苯甲酸雌二醇(estradiol benzoate)是3-位酯,戊酸雌二醇(estradiol valerate)是17β-位酯化衍生物,在合成时先生成双酯,由于3-位酯相对易水解,可部分只保留17β-酯。
estradiol的3或17β位羟基的各种各样的酯化产物是最常见的衍生物,它们能在植物油中溶解制成长效针剂。苯甲酸雌二醇(estradiol benzoate)是3-位酯,戊酸雌二醇(estradiol valerate)是17β-位酯化衍生物,在合成时先生成双酯,由于3-位酯相对易水解,可部分只保留17β-酯。

从药效的观点,3位或17β酯化产物都需要在体内转化成estradiol后才能发生作用,因而选择3位或17β位酯的依据可能主要是考虑合成上的方便。
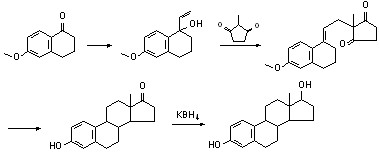
合成路线:estradiol可用全合成法制备:由6-甲氧萘满酮与2-甲基戊二酮-1,3加成,环合及氢化后得到estrone,再用硼氢化钾还原后得本品。
合成路线:estradiol可用全合成法制备:由6-甲氧萘满酮与2-甲基戊二酮-1,3加成,环合及氢化后得到estrone,再用硼氢化钾还原后得本品。
结合雌激素:结合雌激素(conjugated estrogens,商品名premarin)是目前使用较多得雌激素,它是以雌酮硫酸单钠盐、马烯雌酮硫酸单钠盐为主要成分(分别占50%~63%及22.5%~32.5%),尚存在少量17α-雌二醇、马萘雌酮、马萘雌酚及它们的硫酸酯单钠盐,其他所含少量成分是因为天然来源难以分离而夹杂,或在药效学上起协同作用或甚至是因商品化的需要而有意保留,原因不得而知。它是一种口服雌激素药物。该产品是从孕母马尿中提取制得,《美国药典》中收载该产品除天然物外尚有其合成代用品。
estradiol及马烯雌酮均是弱雌激素物质,作为激素替补治疗用药是比较理想的。它们进入体内经硫酸酯化成钠盐后成为水溶性物质而从尿中排泄。因而,本产品实际上是一类代谢产物,是用代谢产物作为药物使用的一种典型实例。
结合雌激素在胃肠道吸收进入体内后再释放出estrone及马烯雌酮而发挥作用,因而在体内是一平衡反应。
estradiol及马烯雌酮均是弱雌激素物质,作为激素替补治疗用药是比较理想的。它们进入体内经硫酸酯化成钠盐后成为水溶性物质而从尿中排泄。因而,本产品实际上是一类代谢产物,是用代谢产物作为药物使用的一种典型实例。
结合雌激素在胃肠道吸收进入体内后再释放出estrone及马烯雌酮而发挥作用,因而在体内是一平衡反应。
二、非甾体雌激素及选择性雌激素受体调节剂
非甾体雌激素主要是二苯乙烯类化合物,选择性雌激素受体调节剂主要是三苯乙烯类化合物。
己烯雌酚 diethylstilbestrol
非甾体雌激素主要是二苯乙烯类化合物,选择性雌激素受体调节剂主要是三苯乙烯类化合物。
己烯雌酚 diethylstilbestrol
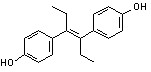
化学名:(E)-4,4′-(1,2-二乙基-1,2-亚乙烯基)双苯酚(4,4′[(1E)-1,2-diethyl-1,2-ethenediyl]bisphenol)。
diethylstilbestrol可以很快从胃肠道吸收,在肝中失活很慢,口服有效,多制成口服片剂应用,也有将它溶在植物油中制成油针剂。
反式己烯雌酚有效,顺式无效,分子中两个苯环取代相对对称,含有两个酚羟基,因而与FeCl3能呈色反应。
diethylstilbestrol的两个酚羟基是活性官能基,用于制备各种衍生物。目前作为商品的最常用的衍生物是己烯雌酚丙酸酯(diethylstilbestrol dipropionate)及己烯雌酚磷酸酯(diethylstilbestrol diphosphate)和它的钠盐。前者作为长效油剂使用。后者主要用于前列腺癌,考虑到癌细胞有较高的磷酸酯酶的活性,药物进入体内后在癌细胞中更易被水解释放出更多的diethylstilbestrol,提高药物的选择性。钠盐可制成静脉注射剂。
diethylstilbestrol可以很快从胃肠道吸收,在肝中失活很慢,口服有效,多制成口服片剂应用,也有将它溶在植物油中制成油针剂。
反式己烯雌酚有效,顺式无效,分子中两个苯环取代相对对称,含有两个酚羟基,因而与FeCl3能呈色反应。
diethylstilbestrol的两个酚羟基是活性官能基,用于制备各种衍生物。目前作为商品的最常用的衍生物是己烯雌酚丙酸酯(diethylstilbestrol dipropionate)及己烯雌酚磷酸酯(diethylstilbestrol diphosphate)和它的钠盐。前者作为长效油剂使用。后者主要用于前列腺癌,考虑到癌细胞有较高的磷酸酯酶的活性,药物进入体内后在癌细胞中更易被水解释放出更多的diethylstilbestrol,提高药物的选择性。钠盐可制成静脉注射剂。
枸橼酸他莫昔芬 tamoxifen Citrate

化学名:(Z)-N,N-二甲基-2-[4-(1,2-二苯基-1-丁烯基)苯氧基]-乙胺的枸橼酸盐((Z)-2-[4-(1,2-diphenyl-1-butenyl)phenoxyl]-N,N-dimethyl ethanamine citrate)。
tamoxifen为三苯乙烯类化合物,以己烯雌酚类雌激素为先导物发展出来的抗雌激素药物。分子中具有二苯乙烯的基本结构,其中双键一端碳上增加二甲氨基乙氧苯基,此取代基是很多药物中的结构单元。按几何异构化学命名原则,由于C-1上取代苯基立体排列顺序大于苯,而在C-2中苯基的序数又大于乙基,故药用品为顺式几何异构体,反式异构体的活性小于顺式。
tamoxifen由于肝肠循环以及和清蛋白高度的结合力,使得半衰期长达7d左右。
口服tamoxifen后,能很好的吸收,在体内被广泛代谢。代谢物主要在胆汁中以结合物形式排泄,只有极少量或没有未被代谢的tamoxifen被排出体外。
tamoxifen为三苯乙烯类化合物,以己烯雌酚类雌激素为先导物发展出来的抗雌激素药物。分子中具有二苯乙烯的基本结构,其中双键一端碳上增加二甲氨基乙氧苯基,此取代基是很多药物中的结构单元。按几何异构化学命名原则,由于C-1上取代苯基立体排列顺序大于苯,而在C-2中苯基的序数又大于乙基,故药用品为顺式几何异构体,反式异构体的活性小于顺式。
tamoxifen由于肝肠循环以及和清蛋白高度的结合力,使得半衰期长达7d左右。
口服tamoxifen后,能很好的吸收,在体内被广泛代谢。代谢物主要在胆汁中以结合物形式排泄,只有极少量或没有未被代谢的tamoxifen被排出体外。
三、雄性激素和蛋白同化激素
雄性激素能促进男性性器官及副性征的发育、成熟,对抗雌激素抑制,抑制子宫内膜生长及卵巢、垂体功能。同时,还具有蛋白同化作用,即促进蛋白质合成和骨质形成,刺激骨髓造血功能,以及蛋白质代谢,从而使肌肉增长,体重增加。对雄性激素的化学结构修饰的结果导致得到一些雄性活性很微弱,而蛋白同化活性增强的新化合物。它们常被称作蛋白同化激素,对雄性激素的化学结构修饰的主要目的就是为了获得蛋白同化激素。雄性活性的结构专一性很强,对testosterone的结构稍加变动(如19去甲基、A环取代、A环骈环等修饰)就可使雄性活性降低及蛋白同化活性增加。但要做到完全没有雄性活性是十分困难的,因此,雄性活性仍是蛋白同化激素的主要副作用。
代表药物 丙酸睾酮 testosterone propionate
雄性激素能促进男性性器官及副性征的发育、成熟,对抗雌激素抑制,抑制子宫内膜生长及卵巢、垂体功能。同时,还具有蛋白同化作用,即促进蛋白质合成和骨质形成,刺激骨髓造血功能,以及蛋白质代谢,从而使肌肉增长,体重增加。对雄性激素的化学结构修饰的结果导致得到一些雄性活性很微弱,而蛋白同化活性增强的新化合物。它们常被称作蛋白同化激素,对雄性激素的化学结构修饰的主要目的就是为了获得蛋白同化激素。雄性活性的结构专一性很强,对testosterone的结构稍加变动(如19去甲基、A环取代、A环骈环等修饰)就可使雄性活性降低及蛋白同化活性增加。但要做到完全没有雄性活性是十分困难的,因此,雄性活性仍是蛋白同化激素的主要副作用。
代表药物 丙酸睾酮 testosterone propionate
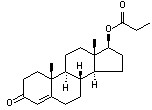
化学名:17β-羟基雄甾-4-烯-3-酮丙酸酯((17β)-17-hydroxyandrost-4-en-3-one propionate)。又名丙酸睾丸素。
发现过程:testosterone是天然雄性激素,1935年从雄仔牛睾丸中提取制得纯品。后经结构阐明为雄甾烷衍生物。在母核上取代有 -3-酮及17β-羟基,本品是其丙酸酯化合物。
-3-酮及17β-羟基,本品是其丙酸酯化合物。
结构改造:作为雄性激素替补治疗药物的天然testosterone,进行结构修饰的目的主要为了使用方便和达到长效。如testosterone除丙酸酯之外,尚有戊酸酯和十一烯酸酯作为长效药物,可每周或每月使用一次。testosterone的17α-甲基衍生物口服吸收很快,生物利用度好,又不易在肝内被破坏。甲睾酮(methyltestosterone)现已作为常用的口服雄激素的主要药物。对肝的毒性是其主要的副作用。
发现过程:testosterone是天然雄性激素,1935年从雄仔牛睾丸中提取制得纯品。后经结构阐明为雄甾烷衍生物。在母核上取代有
结构改造:作为雄性激素替补治疗药物的天然testosterone,进行结构修饰的目的主要为了使用方便和达到长效。如testosterone除丙酸酯之外,尚有戊酸酯和十一烯酸酯作为长效药物,可每周或每月使用一次。testosterone的17α-甲基衍生物口服吸收很快,生物利用度好,又不易在肝内被破坏。甲睾酮(methyltestosterone)现已作为常用的口服雄激素的主要药物。对肝的毒性是其主要的副作用。

合成路线:testosterone propionate及methyltestosterone都可以用去氢表雄酮为原料经Oppenauer氧化及KBH4还原,得到testosterone及双氢睾酮的混合物,其中双氢睾酮用MnO2氧化可转化成testosterone。再用相应的酰酐或酰氯酯化即可得到睾酮酯。若将醋酸去氢表雄酮先用碘化甲基镁Grigner反应,水解后经Oppenauer氧化则得methyltesterone。

四、孕激素
黄体酮(progesterone)及17α-羟基黄体酮(17α-hydroxyprogesterone)是天然来源的孕激素。它们与雌激素共同维持女性生殖周期及女性生理特征。目前孕激素主要用于保护妊娠,它与雌激素配伍用做口服避孕药,也用在雌激素替补治疗中,作为抵消副作用的用药。
发现过程:progesterone是最早发现的天然孕激素,1934年首先从孕妇尿中分离出来,一年后确定其化学结构是-3-酮的C-21-甾体。从化学结构来看progesterone与testosterone甾核的-3-酮是完全一样,仅17β位前者是乙酰基后者是羟基。
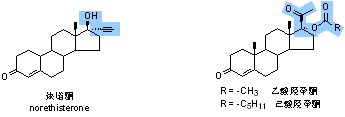
progesterone口服无效,在寻找口服孕激素的研究中,第一个成为口服有效药物的不是progesterone衍生物,而是testostrone衍生物―炔孕酮(ethisterone),17α位引入乙炔基后,雄激素活性减弱而显示出孕激素活性,且口服有效。不久以后,在研究皮质激素生物合成过程中,发现17α-hydroxyprogesterone,它无口服活性,经乙酰化后口服活性增加,虽其口服活性仅有炔诺酮(norethisterone)的1/100,但从此开辟出一类黄体酮类口服孕激素。若用己酸酐进行酰化得己酸羟孕酮(17α-hydroxyprogesterone caproate),为长效孕激素,其油剂注射一次延效1个月。
黄体酮(progesterone)及17α-羟基黄体酮(17α-hydroxyprogesterone)是天然来源的孕激素。它们与雌激素共同维持女性生殖周期及女性生理特征。目前孕激素主要用于保护妊娠,它与雌激素配伍用做口服避孕药,也用在雌激素替补治疗中,作为抵消副作用的用药。
发现过程:progesterone是最早发现的天然孕激素,1934年首先从孕妇尿中分离出来,一年后确定其化学结构是
progesterone口服无效,在寻找口服孕激素的研究中,第一个成为口服有效药物的不是progesterone衍生物,而是testostrone衍生物―炔孕酮(ethisterone),17α位引入乙炔基后,雄激素活性减弱而显示出孕激素活性,且口服有效。不久以后,在研究皮质激素生物合成过程中,发现17α-hydroxyprogesterone,它无口服活性,经乙酰化后口服活性增加,虽其口服活性仅有炔诺酮(norethisterone)的1/100,但从此开辟出一类黄体酮类口服孕激素。若用己酸酐进行酰化得己酸羟孕酮(17α-hydroxyprogesterone caproate),为长效孕激素,其油剂注射一次延效1个月。
代表药物 醋酸甲羟孕酮 medroxyprogesterone acetate
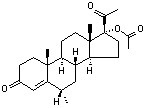
化学名:6α-甲基-17α-羟基孕甾-4-烯-3,20-二酮醋酸酯((6α)-17-hydroxy-6-methylpregn-4-ene-3,20-dione acetate)。
发现过程:醋酸甲羟孕酮(medroxyprogesterone acetate)是17α乙酰氧基黄体酮的6α-甲基取代物。对黄体酮的药物代谢研究发现,孕酮类化合物失活的主要途径是6位羟基化,16位和17位氧化,或3,20二酮被还原成二醇。
因而结构修饰主要是在C6及C16位上进行,如用烷基、卤素、双键等进行取代,结果是满意的。17α-乙酰氧基黄体酮的6α-甲基衍生物,即醋酸甲羟孕酮(medroxyprogesterone acetate)及△6-6-氯衍生物,即醋酸甲地孕酮(megestrol acetate),及△6-6-氯衍生物,即醋酸氯地孕酮(chlormadinone acetate)都是强效口服孕激素,其活性分别是norethisterone的20、12及50倍,也是目前常用的孕激素药物。
发现过程:醋酸甲羟孕酮(medroxyprogesterone acetate)是17α乙酰氧基黄体酮的6α-甲基取代物。对黄体酮的药物代谢研究发现,孕酮类化合物失活的主要途径是6位羟基化,16位和17位氧化,或3,20二酮被还原成二醇。
因而结构修饰主要是在C6及C16位上进行,如用烷基、卤素、双键等进行取代,结果是满意的。17α-乙酰氧基黄体酮的6α-甲基衍生物,即醋酸甲羟孕酮(medroxyprogesterone acetate)及△6-6-氯衍生物,即醋酸甲地孕酮(megestrol acetate),及△6-6-氯衍生物,即醋酸氯地孕酮(chlormadinone acetate)都是强效口服孕激素,其活性分别是norethisterone的20、12及50倍,也是目前常用的孕激素药物。

构效关系:

五、甾体避孕药
代表药物 左炔诺孕酮 levonorgestrel
代表药物 左炔诺孕酮 levonorgestrel
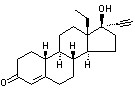
化学名:D-(-)-17α-乙炔基-17β-羟基-18-甲基雌甾-4-烯-3-酮((17α)-ethynyl-17(β)-hydroxy-18-methyl-estro-4-en-3-on)。
发现过程:左炔诺孕酮(levonorgestrel)是在炔诺酮(norethisterone)的基础上发展起来的,norethisterone是第一个上市的19去甲(19nor-)型甾体孕激素,前面已有介绍ethisterone是睾酮的乙炔化物,具有口服孕激素活性,但它们仍保留有testosterone的1/10雄性活性,难以被妇女接受。经过进一步结构修饰,将ethisterone的19位甲基除去后即norethisterone,发现其孕激素活性增大了5倍而雄性活性又小了1倍,可为妇女接受而上市。
临床用途:levonorgestrel的作用及用途与norethisterone一样,能抑制垂体释放LH和SH,抑制排卵作用强于progesterone,但口服后吸收完全,生物利用度极好(87%~99%),比norethisterone(70%)大。其孕激素活性亦比norethisterone强,而抗雌激素活性亦增加,也有一定的雄激素及同化激素化作用。因而本品的药效、药代总体评价比norethisterone有更多优点及更小的副作用,在世界上较广泛的使用。
六、孕激素拮抗剂
孕激素拮抗剂的定义:孕激素拮抗剂指与孕激素竞争受体并拮抗其活性的化合物,也称抗孕激素(antiprogestins)。
发现过程:在20世纪80年代之前抗孕激素尚未成为药物,尽管对抗孕激素的活性及构效关系有许多研究,终因没有找到恰当的适应证,研究工作停滞不前。1982年法国Roussel-Uclaf公司推出米非司酮(mifepristone)作为抗早孕药物,不但促进了抗孕激素及抗皮质激素药的发展,而且在甾体药物研究历史上起着里程碑的作用。它使得已经变得不甚活跃的甾体药物研究领域重新燃起了新的期望。
抗孕激素作用的靶部位是孕激素受体。目前主要用于抗早孕,也有些抗孕激素药物用于乳腺癌的治疗。
发现过程:左炔诺孕酮(levonorgestrel)是在炔诺酮(norethisterone)的基础上发展起来的,norethisterone是第一个上市的19去甲(19nor-)型甾体孕激素,前面已有介绍ethisterone是睾酮的乙炔化物,具有口服孕激素活性,但它们仍保留有testosterone的1/10雄性活性,难以被妇女接受。经过进一步结构修饰,将ethisterone的19位甲基除去后即norethisterone,发现其孕激素活性增大了5倍而雄性活性又小了1倍,可为妇女接受而上市。
临床用途:levonorgestrel的作用及用途与norethisterone一样,能抑制垂体释放LH和SH,抑制排卵作用强于progesterone,但口服后吸收完全,生物利用度极好(87%~99%),比norethisterone(70%)大。其孕激素活性亦比norethisterone强,而抗雌激素活性亦增加,也有一定的雄激素及同化激素化作用。因而本品的药效、药代总体评价比norethisterone有更多优点及更小的副作用,在世界上较广泛的使用。
六、孕激素拮抗剂
孕激素拮抗剂的定义:孕激素拮抗剂指与孕激素竞争受体并拮抗其活性的化合物,也称抗孕激素(antiprogestins)。
发现过程:在20世纪80年代之前抗孕激素尚未成为药物,尽管对抗孕激素的活性及构效关系有许多研究,终因没有找到恰当的适应证,研究工作停滞不前。1982年法国Roussel-Uclaf公司推出米非司酮(mifepristone)作为抗早孕药物,不但促进了抗孕激素及抗皮质激素药的发展,而且在甾体药物研究历史上起着里程碑的作用。它使得已经变得不甚活跃的甾体药物研究领域重新燃起了新的期望。
抗孕激素作用的靶部位是孕激素受体。目前主要用于抗早孕,也有些抗孕激素药物用于乳腺癌的治疗。
米非司酮 mifepristone

化学名:11β-(4-二甲氨基)-17β-羟基-17-(1-丙炔基)-雌甾-4,9-二烯-3-酮((11β,)-[4-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one)。
发现过程:mifepristone的发明与抗雌激素药物的发现是有联系的,tamoxifen及其他的抗雌激素化合物具有三苯基乙烯基的结构,其结构中的双键不同碳上连接的两个苯环,相当于甾体激素分子中的A环和D环,表11-7中的非甾体雌激素diethylstibestrol所示。tamoxifen分子中的第三个苯环所处位置,相当于甾体母核中的β侧的C-11位,这种结构特点对抗激素作用极为重要。
发现过程:mifepristone的发明与抗雌激素药物的发现是有联系的,tamoxifen及其他的抗雌激素化合物具有三苯基乙烯基的结构,其结构中的双键不同碳上连接的两个苯环,相当于甾体激素分子中的A环和D环,表11-7中的非甾体雌激素diethylstibestrol所示。tamoxifen分子中的第三个苯环所处位置,相当于甾体母核中的β侧的C-11位,这种结构特点对抗激素作用极为重要。
七、肾上腺皮质激素
分类:肾上腺皮质激素按其生理作用特点可分为盐皮质激素(mineralcorticoids)及糖皮质激素(glucocorticoids)。
代表药物 氢化可的松 hydrocortisone
分类:肾上腺皮质激素按其生理作用特点可分为盐皮质激素(mineralcorticoids)及糖皮质激素(glucocorticoids)。
代表药物 氢化可的松 hydrocortisone
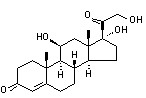
化学名:11β,17α,21-三羟基孕甾-4-烯-3,20-二酮(11β,17α,21-trihydroxy-pregn-4-ene-3,20-dione)。
构效关系:
1.C-21位的修饰 hydrocortisone分子中的三个羟基,用常规方法进行酯化时,如与酸酐或酰氯反应时,只有C-21羟基能被酯化,C-11羟基团,C-13及C-18角甲基的位阻,C-17羟基因侧链的位阻均不能形成酯。
构效关系:
1.C-21位的修饰 hydrocortisone分子中的三个羟基,用常规方法进行酯化时,如与酸酐或酰氯反应时,只有C-21羟基能被酯化,C-11羟基团,C-13及C-18角甲基的位阻,C-17羟基因侧链的位阻均不能形成酯。
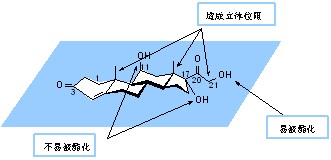
hydrocortisone与醋酐反应,可得C-21位羟基被酯化的前体药物——醋酸氢化可的松(hydrocortisone acetate),其作用时间延长,稳定性增加。之后有一系列酯类衍生物问世。其中,长碳链脂肪酸酯及二元有机酸的单酯钠及磷酸酯盐,它们均为前药,前者水溶性小,常以口服或局部给药,可延长作用时间,后者可制成水溶液供注射用,如氢化可的松琥珀酸钠、氢化可的松磷酸酯钠盐,水溶性大。C-21位的修饰不改变糖皮质激素的活性。
2.C-1位的修饰 以hydrocortisone acetate为先导化合物,经C1-2位脱氢在A环引入双键后得到醋酸泼尼松龙(hydroprednisone acetate),其抗炎活性比其先导物大4倍,而钠潴留作用不变。对这种活性改变的解释是认为A环构型从半椅式变成船式,能提高与受体的亲和力。
C-1位的修饰是对皮质激素甾环母核结构改变的起点,之后,一些强效皮质激素都采用了这一结构修饰手段。
2.C-1位的修饰 以hydrocortisone acetate为先导化合物,经C1-2位脱氢在A环引入双键后得到醋酸泼尼松龙(hydroprednisone acetate),其抗炎活性比其先导物大4倍,而钠潴留作用不变。对这种活性改变的解释是认为A环构型从半椅式变成船式,能提高与受体的亲和力。
C-1位的修饰是对皮质激素甾环母核结构改变的起点,之后,一些强效皮质激素都采用了这一结构修饰手段。
醋酸地塞米松 dexamethasone acetate
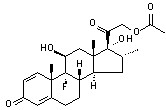
化学名:16α-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3-20二酮-21-醋酸酯((11β,16α)-9-Fluoro-11,17,21-trihydroxy-16-methylpregna-1,4-diene-3,20-dione-21-acetate)。
结构特点:dexamethasone acetate有明显的化学结构特点,在孕甾烷的母核上,几乎在可能被取代的位置上都引入了取代基。如C-1,2及C-4,5的双键,C-3的酮基,C-9的氟、C-11β、C-17α及C-21羟基取代,而且C-16有C-16α-甲基取代。(C-16β-甲基取代也是皮质激素药物——倍他米松)。
结构特点:dexamethasone acetate有明显的化学结构特点,在孕甾烷的母核上,几乎在可能被取代的位置上都引入了取代基。如C-1,2及C-4,5的双键,C-3的酮基,C-9的氟、C-11β、C-17α及C-21羟基取代,而且C-16有C-16α-甲基取代。(C-16β-甲基取代也是皮质激素药物——倍他米松)。

构效关系:糖皮质激素是具有下面基本结构的21个碳甾体激素。糖皮质激素的C-17位有一羟基成酯,C-11位有一氧原子以羟基或羰基氧的形式存在,C-6、C-9、C-16位可发生取代。修饰糖皮质激素的母核将影响药理活性,而且多个部位的修饰比单个部位的修饰更具作用。
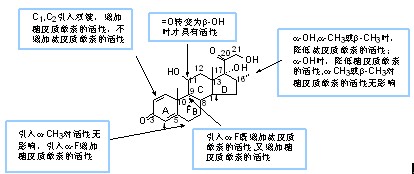
1. C-9位的修饰 对皮质激素类药物C-9位结构的修饰是提高作用强度的不可缺乏的手段,现在强效皮质激素几乎都有C-9F取代。
9α-氟代氢化可的松是最早引人注意的合成皮质激素,它的发现是偶然的。在氢化可的松的合成过程中,引入11-羟基时,同时产生α和β异构体,(现在C-11羟基已用具有立体选择性的微生物法,引入的都是β羟基),为了使无效的α体转为有效的β体,当时设计了几步路线。中间体9-卤化物经药理筛选发现,它们的药理活性比母体化合物大有增加,其中以9α-氟化物作用最强,抗炎活性和糖原沉积活性比氢化可的松大10倍。可惜,由于钠潴留作用增加更多(50倍),最终它未能成为内用药物,只能作为外用皮肤病治疗药,然而却鼓励人们去寻找只增加抗炎活性而不增加钠潴留作用的新药。
2. C-16位的修饰 后来发现在C-9引入氟的同时再在C-16上引入基团可消除钠潴留的作用。在患肾上腺癌病人的尿中发现hydrocortisone的16α羟基代谢产物,它的糖皮质激素活性依旧保留,而钠潴留的副作用明显降低。从代谢产物中寻找新的药物是人们一贯常用的手段,这里也不例外。因而在C-9氟甾体中对引入甲基进行了研究。发现C-16甲基的引入使17α羟基及C-20羰基在血浆中的稳定性增加,其抗炎活性比hydrocortisone大20倍、抗风湿性大30倍。
3. C-6位的修饰 在C-6位引入氟原子后可阻滞C-6氧化失活,如醋酸氟轻松(fluocinonide acetate),其抗炎及钠潴留活性均大幅增加,而后者增加得更多,因而只能外用,治疗皮肤过敏症。
9α-氟代氢化可的松是最早引人注意的合成皮质激素,它的发现是偶然的。在氢化可的松的合成过程中,引入11-羟基时,同时产生α和β异构体,(现在C-11羟基已用具有立体选择性的微生物法,引入的都是β羟基),为了使无效的α体转为有效的β体,当时设计了几步路线。中间体9-卤化物经药理筛选发现,它们的药理活性比母体化合物大有增加,其中以9α-氟化物作用最强,抗炎活性和糖原沉积活性比氢化可的松大10倍。可惜,由于钠潴留作用增加更多(50倍),最终它未能成为内用药物,只能作为外用皮肤病治疗药,然而却鼓励人们去寻找只增加抗炎活性而不增加钠潴留作用的新药。
2. C-16位的修饰 后来发现在C-9引入氟的同时再在C-16上引入基团可消除钠潴留的作用。在患肾上腺癌病人的尿中发现hydrocortisone的16α羟基代谢产物,它的糖皮质激素活性依旧保留,而钠潴留的副作用明显降低。从代谢产物中寻找新的药物是人们一贯常用的手段,这里也不例外。因而在C-9氟甾体中对引入甲基进行了研究。发现C-16甲基的引入使17α羟基及C-20羰基在血浆中的稳定性增加,其抗炎活性比hydrocortisone大20倍、抗风湿性大30倍。
3. C-6位的修饰 在C-6位引入氟原子后可阻滞C-6氧化失活,如醋酸氟轻松(fluocinonide acetate),其抗炎及钠潴留活性均大幅增加,而后者增加得更多,因而只能外用,治疗皮肤过敏症。