第十章 缺血―再灌注损伤
概述
※缺血―再灌注损伤(ischemia―reperfusion injury IRI): 血液再灌注后缺血性损伤进一步加重的现象。【重点】
氧反常(oxygen paradox):用低氧溶液灌注组织器官或在缺氧条件下培养细胞一定时间后,再恢复正常氧供应,组织及细胞损伤不仅未能恢复,反而更趋严重的现象。
钙反常(calcium paradox):预先用无钙溶液灌流大鼠心脏2min,再用含钙溶液进行灌流时,心肌细胞酶释放增加,肌纤维过度收缩及心肌电信号异常。
pH反常( pH paradox):在再灌注时迅速纠正缺血组织的酸中毒,反而会加重缺血―再灌注损伤。
这提示氧、钙和PH可能参与缺血―再灌注损伤的发生、
发展。
第一节 缺血―再灌注损伤的原因及条件
一、原因
1、组织器官缺血后恢复血液供应,如休克时微循环的疏通,冠状动脉痉挛的缓解,心脏骤停后心脑肺复苏等。
2、动脉搭桥术,冠脉血管成形术(PTCA)、溶栓疗法等血管再通术后,心脏外科体外循环术,器官移植及断肢再植术后。
二、影 响 因 素
1、缺血时间
2、侧枝循环
3、需氧程度
4、再灌注条件
第二节 缺血―再灌注损伤的发生机制
一、 自由基的作用
(一) 自由基的概念与类型 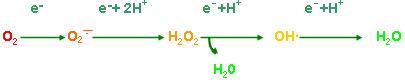
分子氧在线粒体细胞色素氧化酶系统中,获得一个电子还原生成超氧阴离子(O2),获得2 个电子生成H2O2,获得3 个电子生成羟自由基(OH),获得4个电子还原生成H2O,同时释放能量。
※自由基(free radial,FR):是在外层电子轨道上含有单个不配对电子的 原子、原子团和分子的总称。【重点】
自由基的种类很多,主要包括:
※ 1、氧自由基(oxygen free radical, OFR):由氧诱发的自由基。包括O2、OH'。 属非酯性自由基。
※ 2、活性氧(reactive oxygen,RO): 为化学性质较基态氧活泼的含氧物质,包括氧自由基和非自由基的含氧产物,如 H2O2,1O2。
单线态氧(1O2):是一种激发态氧,易氧化不饱和脂肪酸。

2、脂性自由基:指氧自由基与多价不饱和脂肪酸作用后生成 的中间代谢产物,如烷自由基(L?),烷氧自由基(LO?)烷过氧自由基( LOO? )等 。
3、其它:氯自由基(CI?)、甲基自由基(CH3? )和NO等自由基的性质极为活泼,易于失去电子而氧化,或获得电子而还原,特别是其氧化作用很强,故具有强烈的引发脂质化作用。

※(二)氧自由基生成增多的机制【重点】
1.黄嘌呤氧化酶的形成增多
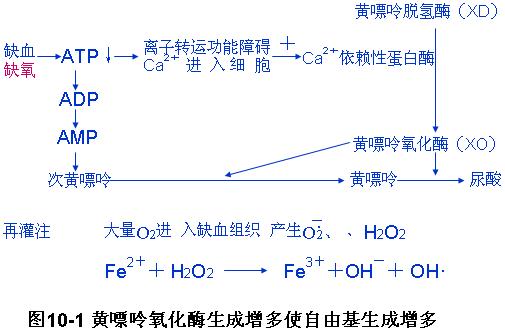
XO及其前身黄嘌呤脱氢酶(XD)主要存在于毛细血管内皮细胞内。正常情况下,90%以XD的形式存在,XO仅占10%。当组织缺血缺氧时,由于ATP含量降低,离子转运功能障碍,细胞外钙离子进入细胞内,激活了钙离子依赖性蛋白酶,使XD转变成XO。同时,由于ATP分解,ADP和AMP含量升高,并依次分解成次黄嘌呤,造成次黄嘌呤大量堆积。再灌注时,大量分子氧随血液进入缺血组织,XO在催化次黄嘌呤并进而催化黄嘌呤生成尿酸的过程中释放出大量的电子,为分子氧所接受,产生O2-.和H2O2。
2、中性粒细胞 中性粒细胞在吞噬活动时耗氧量增加,组织缺血可以激活补体系统,或经细胞膜分解产生多种具有趋化活性的物质,如C3片段、白三烯等,吸引、激活中性粒细胞。再灌注时组织重新获得O2供应,激活的中性粒细胞耗氧量显著增加,产生大量的氧自由基。
※再灌注期组织重新获得O2供应,激活的中性粒细胞耗氧量显著增加,产生大量氧自由基,称呼吸爆发或氧爆发(respiratory or oxygen burst)。【重点】
3、线粒体 缺血缺氧时,使ATP含量减少,钙进入线粒体增多,使线粒体功能受损,细胞色素C氧化酶系统功能失调,以致进入细胞内的氧经单电子还原而形成的氧自由基增多,而经4价还原生成的水减少。
4、儿茶酚胺的自身氧化 缺血缺氧可刺激机体释放大量儿茶酚胺,过量的儿茶酚胺特别是它的氧化产物可产生具有细胞毒性的氧自由基。
※(三)自由基的损伤作用
★《缺血再灌注损伤时自由基损伤作用的机制》〖重点、难点〗
自由基可与各种细胞成分,如膜磷脂、蛋白质、核酸等发生反应,造成细胞结构损伤和功能障碍。
(1)膜脂质过氧化增强
①破坏膜的正常结构 脂质过氧化使膜不饱和脂肪酸减少,不饱和脂肪酸/蛋白质的比例失调,使膜的液态性、流动性降低,通透性增加,细胞外Ca2+内流增加。
②间接抑制膜蛋白的功能 脂质的过氧化反应使膜脂质发生交联和聚和,可间接抑制膜蛋白的功能,如钙泵、钠泵及Na+/Ca2+交换系统等功能,导致胞浆Na+,Ca2+浓度升高,造成细胞肿胀和钙超载。膜液态性的降低和膜成分的改变影响信号转导分子在膜上和膜内的移动,抑制受体、G蛋白与效应器的偶联,造成细胞信号转导障碍。
③促进自由基和其它生物活性物质生成 脂质过氧化可激活磷脂酶C、磷脂酶D,进一步分解膜磷脂,催化花生四烯酸代谢反应,在增加自由基生成和脂质过氧化的同时,形成多种生物活性物质,如前列腺素、血栓素、白三烯等,吸引、激活中性粒细胞,生成自由基和引起微循环障碍,出现无复流现象,促进再灌注损伤。
④减少ATP生成 线粒体膜脂质过氧化,导致线粒体功能抑制,ATP生成减少,细胞能量代谢障碍。
(2)蛋白质功能抑制 自由基可使酶的巯基氧化,形成二硫键。也可使氨基酸残基氧化,胞浆及膜蛋白和某些酶的交联形成二聚体或更大的聚合物,直接抑制蛋白质的功能。膜离子通道蛋白的抑制与膜磷脂微环境的改变一起,共同导致膜离子梯度异常。自由基可损伤肌纤维蛋白,抑制心肌收缩力。肌浆网钙转运蛋白受自由基的损伤,钙调节功能异常。
(3)、破坏核酸及染色体 自由基可使碱基羟化或DNA断裂,从而引起染色体畸变或细胞死亡。
二、钙超载
※ 钙超载(Calcium overload):各种原因引起的细胞内钙含量异常增多并导致细胞结构损伤和功能代谢障碍的现象。【重点、】 严重者可造成细胞死亡。
〔Na+/ Ca2+交换〕 Na+/ Ca2+交换属逆向离子交换,是通过Na+/Ca2+交换蛋白的变构调节实现的。 Na+/ Ca2+交 换蛋白以3个Na+交换1 个Ca2+的比例对细胞内外Na+、Ca2+进行双相转运。 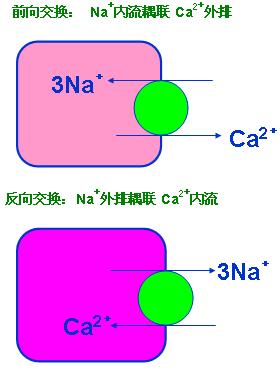
细胞呈何种交换形式主要与的跨膜电化学梯度有关。生理条件下, Na+/ Ca2+交换蛋白主要的转运方向是前向交换将细胞内Ca2+运出细胞与细胞膜钙泵共同维持心肌细胞静息状态的低钙浓度。Na+/ Ca2+交换不直接消耗能量,亦无ATP酶参与,其所需的能量来源于膜上ATP依赖酶( Na+―K+ATP酶和Ca2+ ― Mg2+ ATP酶)活动所提供的Na+和Ca2+的跨膜电化学梯度,属于继发性主动转运。
★ (一)细胞内钙超载的机制【重点、难点】
(1)、Na+/Ca2+ 交换异常 Na+/Ca2+ 交换蛋白是缺血-再灌注损伤和钙反常时大量Ca2+ 进入细胞的主要途径。
①细胞内高Na+直接对Na+/Ca2+ 交换蛋白的激活 缺血使细胞内ATP含量减少,钠泵活性降低,造成细胞内钠含量增高。再灌注时,缺血细胞重新获得氧及营养物质供应,细胞内高Na+除激活钠泵外,还迅速激活的Na+/Ca2+ 交换蛋白,以加速Na+向细胞外转运,同时将大量钙运入胞浆。
②细胞内细胞内高H+对Na+/Ca2+ 交换蛋白的间接激活 缺血期,由于无氧代谢使H+生成增多,组织间液和细胞内pH明显降低。血液再灌注使组织间液H+浓度迅速下降,而细胞内H+仍然很高,形成跨膜H+浓度梯度。细胞膜两侧H+浓度差可激活心肌H+/Na+交换蛋白,促进细胞内H+排出,而使细胞外Na+内流。如果内流的Na+不能被钠泵充分排出,细胞内高Na+就可继发性激活Na+/Ca2+蛋白,促进Ca2+内流,加重细胞钙超载。
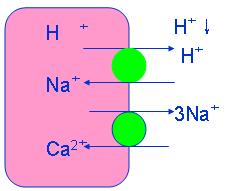
③蛋白激酶C(PKC)对Na+/Ca2+ 交换蛋白的间接激活:

G蛋白-磷脂酶C(PLC)介导的细胞信号转导通路,促进磷酯酰肌醇分解,生成甘油二酯(DG)和三磷酸肌醇(IP3)。DG激活PKC促进Na+/H+交换,进而增加Na+/Ca2+,使胞浆Ca2+浓度升高。IP3促进Ca2+释放。
β肾上腺素能受体兴奋可通过增加L型(受体领带性钙通道)钙通道的开放促进Ca2+内流。钙通道可能也是细胞外Ca2+ 进入胞浆造成钙超载的途径之一。
(2)、生物膜损伤
①细胞膜损伤:再灌注时,自由基的大量生成,使细胞膜脂质过氧化化,加重膜结构的破坏。
②线粒体及肌浆网膜损伤:自由基损伤和膜磷酯分解可造成肌浆网损伤,钙泵功能抑制使肌浆网摄Ca2+ 减少,胞浆钙浓度升高。线粒体损伤抑制氧化磷酸化,ATP生成减少,细胞膜和肌浆网钙泵能量供应不足,促进钙超载的发生。
▲(二)钙超载引起缺血―再灌注损伤的机制【难点】
(1)线粒体功能障碍:在灌注早期,胞浆内Ca2+ 浓度增加,刺激线粒体钙泵(钙-ATP酶)摄钙,线粒体过多摄入钙,消耗了大量ATP的同时,Ca2+与线粒体内含磷酸根的化合物结合,形成不溶性磷酸钙,干扰线粒体的氧化磷酸化,使ATP生成减少。
(2)激活磷脂酶:细胞内钙浓度升高可激活多种磷脂酶,促进膜磷脂分解,损伤细胞膜和细胞器膜。此外,膜磷脂降解产物花生四烯酸、溶血磷脂等增多,亦可加重细胞功能紊乱。
(3)心律失常 通过Na+/Ca2+交换形成一过性内向离子流,在心肌动作电位后形成迟后除极。而导致心律失常的发生。
(4)促进氧自由基的生成 细胞内Ca2+增加可通过增强钙依赖性蛋白酶活性,加速黄嘌呤脱氢酶(XD)转化为黄嘌呤氧化酶(XO),从而促进氧自由基生成。
(5)肌原纤维过度收缩,断裂:①缺血后再灌注使缺血细胞重新获得能量,在胞浆存在高浓度钙的条件下,肌原纤维发生过度收缩。这种肌原纤维过度收缩甚至是不可逆性缩短可损伤细胞骨架结构,造成心肌纤维断裂。②缺血-再灌注使缺血期堆积的H+迅速移出,减轻或消除了H+对心肌收缩的抑制作用。
二、 微血管损伤和白细胞的作用
(一)再灌注时血管内皮细胞与白细胞激活
粘附分子(adhesion molecule,AM):是指由细胞合成的,可促进细胞与细胞之间,细胞与细胞外基质之间粘附的一大类分子的总称。如整合素、选择素、细胞间粘附分子、血管细胞粘附分子及血小板内皮粘附分子等,在维持细胞结构完整和细胞信号转导中起重要作用。
血管内皮细胞激活表现在:在缺血―再灌注早期,血管内皮细胞内原先储存的一些蛋白质前体活化,释放多种细胞粘附分子,促进中性粒细胞粘附和聚集;在再灌注数小时后,血管内皮细胞内一些蛋白质在转录水平上表达增加,大量合成细胞粘附分子等。再灌注损伤可使细胞膜磷酯降解,花生四烯酸代谢产物增多,其中有些物质,如白三烯具有很强的白细胞趋化作用,能吸引大量中性粒细胞粘附于血管内皮细胞并进入组织。中性粒细胞与血管内皮细胞粘附后进一步激活,自身合成释放多种具有趋化作用的炎性介质,使白细胞侵润进一步加重。
(二)血管内皮细胞与中性粒细胞介导的缺血―再灌注损伤
※ 无复流现象(no-reflow phenomenon):结扎狗冠状动脉造成心肌局部缺血后,再开放结扎动脉,重新恢复血流,部分缺血区并不能得到充分的血液灌注现象。【重点】
激活的血管内皮细胞与白细胞相互作用,对微血管和细胞损伤产生的重要影响,是无复流现象的病理生理基础。
◆ 《激活的血管内皮细胞和中性粒细胞在缺血再灌注损伤中的作用,即缺血再灌注损伤时,激活的血管内皮细胞与白细胞对微血管和细胞损伤产生的重要影响》【难点】
(1)微血管损伤
①微血管血液流变学改变:在心肌缺血和再灌注早期,即可见中性粒细胞粘附在血管内皮细胞上。随后,有血小板沉积和红细胞缗线状聚集,造成毛细血管阻塞。再灌注期,激活的血管内皮细胞和中性表面可表达选择素,增强血管内皮与白细胞之间的亲和力,使白细胞在微血管内流速减慢,造成“滚动”现象。粘附分子表达进一步增加,中性粒细胞与内皮细胞发生粘附,粘附的中性粒细胞可释放化学趋化介质,如LTs、TXA2、PAF等,加重细胞间粘附和微血管堵塞。
②微血管口径的改变:再灌注时,损伤的血管内皮细胞肿胀,可导致管腔狭窄,阻碍血液灌流。激活的血管内皮细胞和中性粒细胞可释放大量缩血管物质,如ET、ATII、TXA2等,而扩血管物质的合成与释放减少,造成微血管舒缩功能改变。缺血细胞肿胀使微血管受压,也可促进无复流现象的发生,加重细胞的损伤和坏死。
③微血管通透性增高:自由基损伤和中性粒细胞粘附是微血管通透性增高的主要原因。微血管通透性增高使细胞间质水肿。而中心粒细胞从血管内渗出并粘附于心肌细胞,直接释放细胞毒性介质到心肌细胞表面造成其损伤。
(2)细胞损伤
激活的血管内皮细胞与中性粒细胞释放的大量生物活性物质,如自由基、蛋白酶、细胞因子等,不但可改变自身的结构和功能,而且使周围组织细胞受到损伤。 第三节 缺血―再灌注损伤时机体的功能及代谢变化
一、心脏缺血―再灌注损伤的变化
(一)心功能变化
1、缺血―再灌注心律失常:在心肌缺血―再灌注过程中出现的心律失常称为再灌注性心律失常。其中以室性心律失常,如室性心动过速和心室颤动最为多见。
缺血―再灌注心律失常的发生与缺血―再灌注前心肌缺血的时间长短有关。缺血时间过短,心肌损伤不明显;缺血时间过长,心肌丧失电活动,二者均不易出现缺血―再灌注性心律失常。
此外,心律失常的发生还与缺血心肌的数量,缺血和程度,再灌注血流的速度及电解质紊乱等因素有关。
缺血一再灌注性心律失常的发生是多因素的,自由基和钙超载造成的心肌损伤及ATP减少使ATP敏感性钾通道激活等均可改变心肌电生理特性。
2 心肌舒缩功能降低
缺血一再灌注导致的心肌可逆性或不可逆性损伤均造成心肌舒缩功能降低,表现为心输出量减少,心室内压最大变化速率(±dp/dtmax)降低,左室舒张末期压力(LVEDP)升高等。
※ 心肌顿抑(myocardial stunning):心肌并未因缺血发生不可逆损伤,但在再灌注血流已恢复或基本恢复正常后一定时间内心肌出现的可逆性收缩功能降低的现象。【重点】
心肌顿抑是缺血一再灌注损伤的表现形式之一,自由基爆发性生成和钙超载是心肌顿抑的主要发病机制。
(二)心肌代谢变化
缺血期心肌ATP及CP含量降低,ADP、AMP及其降解产物含量升高。如缺血损伤轻、心肌获得O2和代谢底物后,心肌高能磷酸化合物含量可较快恢复正常。如缺血时间较长,再灌注后心肌高能磷酸化合物含量不仅不回升,反而进一步降低。这是因为再灌注时自由基和钙超载对线立体的损伤使心肌能量合成减少;加上再灌注血流的冲洗,ADP、AMP等物质含量比缺血期降低,造成高能磷酸化合物的底物不足。
(三) 心肌超微结构的变化
缺血一再灌注损伤时,心肌超微结构变化较单纯缺血时进一步加重,表现为细胞膜破坏,线粒体肿胀,嵴断裂,溶解,空泡形成,由于Ca2+蓄积,基质内致密颗粒增多,肌原纤维断裂,节段性溶解和出现收缩带。缺血一再灌注还可造成不可逆性损伤,出现心肌出血、坏死。
二、脑缺血-再灌注损伤的变化
(一) 脑缺血-再灌注损伤时细胞代谢的变化
脑缺血后短时间内ATP、CP、葡萄糖等均减少,乳酸明显增加。缺血期cAMP含量增加,而cGMP含量减少。缺血-再灌注后脑内cAMP进一步增加,cGMP进一步下降,这提示缺血-再灌注时脂质过氧化反应增强。 
再灌注后cAMP升高可导致磷脂酶激活,使膜磷脂降解,游离脂肪酸增多,最显著的是花生四烯酸(AA)及硬脂酸增多(SA)自由基与游离脂肪酸作用使过氧化脂质生成增多。

脑缺血时脑细胞生物电发生改变:出现病理性慢波,缺血一定时间后再灌注,慢波持续并加重。
(二)脑缺血一再灌注损伤时组织学变化
脑最明显的组织学变化是脑水肿及脑细胞坏死。脑水肿的产生是膜脂质过氧化使膜的结构破坏和钠泵功能障碍的结果。
三 、 其它器官缺血一再灌注损伤的变化
肠缺血一再灌注损伤
肾缺血一再灌注损伤
骨骼肌缺血一再灌注损伤
第四节 防治缺血一再灌注损伤的病理生理基础
一、 减轻缺血性损伤,控制再灌注条件
二、改善缺血组织的代谢
三、 清除自由基
机体对抗自由基损伤的防护系统主要有两大类:
(一)低分子清除剂
1、存在于细胞脂质部分的自由基清除剂:维生素E和维生素A等。
2、存在于细胞内外水相中的自由基清除剂:半胱氨酸,抗坏血酸,GSH和NADPH等。上述自由基清除剂,能提供电子使自由基还原:
①维生素E能还原O2,1O2脂质自由基等。
②抗坏血酸具有协同作用,而且能协助维持维生素E在具有活性的还原状态。
③维生素A是1O2的有效清除剂并能抑制脂质过氧化。
④胞浆中的GSH与NADPH在过氧化氢酶(CAT)、谷胱甘肽过氧化物酶(GSH―PX)等抗氧化酶的协同作用下,能还原H2O2,过氧化脂质、二硫化物及某些自由基。
(二)酶性清除剂
1、CAT及过氧化物酶存在于细胞内,可以清除H2O2,以避免高毒性OH?的产生。 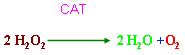
2 、※超氧化物岐化酶(Superoxide dismutase,SOD) 是一种金属蛋白,可以岐化O2生成H2O2。【重点】

SOD作用的重要意义在于清除H2O2和 OH? 的前身O2 ,从而保护细胞不受毒性氧自基的损伤。
四、减轻钙超载
五、其它 |
