第十六章 肾功能不全
第一节 急性肾功能不全
※急性肾功能不全(acute renal insufficiency,ARI):是由于肾小球滤过率急剧减少,或肾小管发生变性坏死而引起的一种严重的急性病理过程,往往出现少尿以及随之而产生的氮质血症、高钾血症,代谢性酸中毒等综合症。【重点】
一、病因与类型
以肾脏为中心,将起ARI的原因分为三类。
(一)肾前因素
凡能引起有效循环血量减少,心输出量下降及引起肾血管收缩的因素,都会导致肾灌流不足,以致GFR下降,而发生急性肾功能不全,称肾前性急性肾功能不全。如各类休克,严重脱水,急性心力衰竭等。
肾前性急性肾功能不全,由于肾尚无器质性病变,一旦肾灌流量恢复,肾功能也恢复正常,属于功能性肾功能衰竭。但持续性肾缺血导致肾小管坏死,则成为器质性肾功能衰竭。
(二)肾性因素
包括肾本身的一些器质性病变和肾缺血,肾毒物引起的急性肾小管坏死等。称为肾性急性肾功能不全。
1、广泛性肾小管损伤:急性肾小球肾炎,全身性红斑狼疮(SLE)。
2、急性肾小管坏死:重金属中毒;药物中毒;生物性毒素等。
3、体液因素异常:低钾血症、高钙血症、高胆红素血症。
4、异型输血:血红蛋白性肾病。
肾性急性肾功能不全是由于肾实质性病变而产生的,属于器质性肾功能衰竭。
(三) 肾后因素
见于肾盂到尿道的尿路急性梗阻。常见的梗阻因素是结石,前列腺肥大,泌尿道周围肿物压迫引起的急性肾功能不全,称肾后性急性肾功能不全。 肾后性ARI早期是功能性的,晚期有肾实质破坏是器质性的。
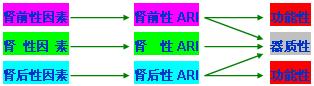
急性肾功能不全根据起病时尿量多少可分为两种类型,即少尿型和非少尿型。
二、少尿型急性肾功能不全
(一)发病经过
1、少尿期 在致病因素作用下,肾血管持续收缩,肾实质缺血,GFR极度降低。开始时肾并无形态改变,如致病因素得不到纠正,出球小动脉持续收缩,终将导致肾小管上皮细胞缺血缺氧而发生变性坏死。此外,肾毒素也可引起急性肾小官坏死(ANT)。临床表现:少尿,无尿。
※24h尿量<400mI为少尿【重点】。
※24h尿量<100mI为无尿【重点】。
由于少尿、无尿、代谢产物蓄积,面出现氮质血症,高钾血症,代谢性酸中毒和水中毒等。
少尿期持续8-16天,一般到第21天进入多尿期。
2、多尿期 当肾小管坏死得到及时而正确的治疗后,坏死的肾小管得以再生与修复,随之功能亦逐渐恢复。此期病人尿量逐渐增多,昼夜排尿3-5L。
多尿的机制:
① 新再生的肾小管上皮细胞的浓缩功能尚未恢复。
② ②蓄积大量的尿素,致使肾小球滤出的尿素增多,肾小管 腔内渗透压升高,引起渗透性利尿。
③由于肾间质水肿消退,肾小管阻塞被解除。
3、恢复期 一般在发病后一个月左右进入恢复期,此时尿液成分大体恢复正常,但肾功能完全恢复则需要更长时间,约3个月至1年。
(二) 少尿的发生机制
少尿发生机制的关键是GFR的降低,影响GFR降低的主要发病机制是:
1、肾缺血
★《急性肾功能不全初期肾缺血的主要机制》【重点】
肾缺血是ARI初期的主要发病机制。造成肾缺血主要与肾灌注压降低,肾血管收缩和肾血液流变学变化有关。
(1) 肾灌注压下降
①肾前性ARI全身血压常低于10.7kPa,使肾灌注压降低,肾小球毛细血管血压下降,导致肾小球有效滤过压下降,使 GFR降低。
②肾后性ARI是由于尿路梗阻引起肾小球囊内压增加,当囊内压和血浆胶体渗透压之和超过肾小球毛细血管血压时,肾小球有效滤过压也可降到零,使GFR降低。
(2)肾血管收缩
在全身血容量降低,肾缺血时,引起肾入球小动脉收缩。入球小动脉收缩的后果是影响GFR,甚至在RBF正常的情况下也引起GFR下降,以及相应肾单位的肾小管缺血。
★《ARI早期肾血管收缩的机制及其影响》【重点】
①体内儿茶酚胺增加:休克或创伤引起的ARI,体内CA浓度急剧增加。皮质肾单位的入球动脉对CA敏感,因而皮质呈缺血改变。
②肾内RAS激活:
1)缺血时肾灌注压降低,刺激近球细胞分泌肾素。
2)近曲肾小管和髓襻升支受损,这些部位对Na+、Cl-重吸收减少,原尿到致密斑Na+浓度升高,刺激致密斑分泌肾素。使肾素-血管紧张素系统激活,血管紧张素Ⅱ增加,引起肾小动脉痉挛,导致GFR降低。
缺血中毒→近曲小管、mTAL受损→对Na+、Cl-主动重吸收↓
→致密斑原尿Na+浓度↑→近球细胞分泌肾素↑→血管紧张素Ⅱ↑→肾入球动脉收缩→ GFR↓
③前列腺素产生减少:肾产生具有扩血管作用PGE2和PGA2减少。
④其它
1)在ARI初期,可见肾内腺苷增多,通过血管壁相应受体,收缩入球小动脉和扩张出球小动脉,使GFR下降。
2) ARI时肾小管细胞内CA2+增多,使肾血管收缩和肾血流量下降。
上述因素主要引起入球小动脉收缩,直接的影响就是导致GFR下降,甚至在RBF正常的情况下也可使GFR下降,以及相应肾单位的肾小管缺血。
(3)血液流变学的变化
①血液粘度升高:ARI时血粘度升高,引起血粘度升高的因素很复杂,其中纤维蛋白原增高可能是使血粘度升高的主要原因。全血粘度升高和入球动脉收缩引起肾小球前阻力增高,影响了肾小球毛细血管床的微循环状态,此时尽管肾灌流正常,但因肾小球毛细血管血压降低,GFR仍下降。
②白细胞在微血管灌注中的作用:短暂肾缺血后,白细胞阻塞微血管,增加血流阻力和降低血流量。
③微血管改变:在ARI时,肾微血管变化主要表现在;血管口径缩小,自动调节功能丧失与血红蛋白附壁。这些变化使肾微血管痉挛,增厚,进一步加剧肾缺血。
2、肾小管阻塞的损伤作用
在缺血性的急性肾小管坏死及异型输血、挤压综合症。磺胺结晶等引起的急性肾小管坏死时,脱落的上皮细胞碎片,肌红蛋白,血红蛋白等所形成的管型阻塞肾小管腔,从而使管腔内压升高,造成肾小球有效滤过压降低而发生少尿。
3、肾小管原尿反流
ARI时,肾小管上皮细胞广泛坏死,基膜断裂,尿液经断裂的基膜返流到肾间质,使间质水肿,并压迫肾小管和肾小管周围的毛细血管。肾小管受压,使阻塞加重。毛细血管受压,血流更进一步减少。肾损害加重,形成恶性循环。
(三)少尿期的代谢紊乱
1、氮质血症 常用血尿素氮(BUN)作为氮质血症指标。ARI不全时,不但不能有效的排出蛋白质代谢产物,而且由于原始病因作用,组织分解增加,使其生成增多,因此血非蛋 白氮增高,发生氮质血症。BUN明显升高是ARI病情严重的一个指标。
2、代谢性酸中毒
3、水中毒 水中毒主要由于:①肾排水减少;②ADH分泌增多;③体内分解代谢增强,内生水增多,细胞水肿,急性肺水肿和脑水肿,稀释性低钠血症。
4、高钾血症 少尿期最危险的并发症 原因:①钾排出减少②组织分解代谢增强,钾从细胞释出;③酸中毒使钾从细胞内向细胞外转移:④低血钠时,肾小球滤液中钠减少,使远曲小管中钾与钠交换随之减少。
(四)肾细织细胞损伤及其机制
▲《肾内各种细胞受损而出现的代谢、功能以及形态结构的紊乱是ARI和GFR持续降低的基本机制》【难点】
1、 受损细胞
(1)肾小管细胞 ARI时肾小管损伤的病理特征有两种:
①小管破裂性损伤:表现为肾小管上皮细胞坏死,脱落,基底膜也被破坏。虽然肾小管各段都可受累,但并不是每个肾单位都会出现损伤。这种损伤在肾中毒及持续性肾缺血病例均可见到。
② 肾毒性损伤:表现为主要损伤近曲小管,可累及所有肾单位,肾小管细胞呈大片状坏死,但基膜完整,主要见于肾中毒的病例。
肾缺血和肾中毒时对肾小管上皮细胞的损伤多表现为细胞功能紊乱而不是坏死。如果细胞坏死或出现形态结构的病理改变,表明损伤已十分严重。
(2)内皮细胞 内皮细胞受损时结构与功能异常包括:
①内皮细胞肿胀,血管管腔变窄,血流阻力增加,肾血流减少。髓质血管内皮细胞的肿胀引起直小血管的血流阻力增加,甚至导致外髓质淤血,缺氧以致肾小管功能、形态结构破坏,可能在ARI时对GFR的持续降低起着重要作用。
②内皮细胞受损激发血小板聚集与微血栓形成以及毛细血管内凝血。肾小球毛细血管内的血栓形成,纤维蛋白沉积具有促进GFR减少的作用。
③肾小球内皮细胞窗变小,甚至减少也可直接影响肾小球超滤系数(kf)使GFR降低。
④内皮细胞释放舒血管因子减少,而释放缩血管因子增多均可加强肾血管的持续收缩,使GFR降低。
(3)系膜细胞:
缺血或中毒性肾损伤时有许多内源性及外源性的活性因子释放如ATⅡ,ADH等,这些物质多数可引起系膜细胞收缩。庆大霉素,腺苷,硝酸铀等毒物也可直接导致肾小球血管阻力增加及肾小球滤过面积改变和kf降低,从而促进GFR持续降低。
▲2、肾组织细胞损伤的机制【难点】
(1)ATP产生减少:缺血时因缺氧及代谢底物缺乏而导致ATP产生减少,缺血及酸中毒引起的线粒体功能障碍也可导致ATP生成障碍。
①ATP减少不仅减弱肾小管的主动重吸收功能,而且由于Na+-K+-ATP酶的活性减弱,细胞内Na+、H2O潴留,发生细胞水肿。
②CA2+-ATP酶活性减弱,使肌浆网摄取CA2+受限及细胞内钙泵出减少,引起细胞浆内游离钙增加。
③随着细胞水肿的发生,细胞膜通透性改变,大量CA2+涌入细胞内,形成细胞内钙超载。细胞内游离CA2+增加又可妨碍线粒体的氧化磷酸化功能,使ATP生成更加减少,从而形成恶性循环最终导致细胞死亡。
④由于缺氧时大量增加的ATP可由线粒体进入胞浆并直接抑制Na+-K+-ATP酶的活性,而且肾毒物也可直接使Na+-K+-ATP酶活性减弱,这更加重了细胞内Na+、水潴留及细胞水肿,妨碍细胞的代谢与功能。
(2)自由基产生增多与清除减少:肾缺血与缺血后再灌注均可使自由基的产生增多。由于缺血引起内源性自由基清除系统代谢底物,SOD等缺乏而使自由基的清除减少,组织与细胞内自由基增加。此外,有些毒物亦可促进自由基的产生,肾毒性免疫性损伤时白细胞可释放大量的自由基。自由基在组织与细胞内增高可引起各种细胞损伤。
(3)还原型谷胱甘肽(GSH)减少:肾缺血及肾中毒时,肾组织中GSH显著减少,使细胞抗氧化与过氧化的能力减弱,从而CA2+浓度明显升高以及GSH显著降低的情况下,磷脂酶A2(PLA2)活性增高,因而不仅大量释放脂肪酶,而且使细胞骨架结构解体,各种膜被降解。大量的脂肪酸如AA,还可分解产生PGS,LTS等产物,从而影响血管张力,血小板聚集以及肾小管上皮细胞的功能。
三、非少尿型急性肾功能不全
非少尿型ARI虽然尿量不减少甚至增多,因GFR降低同样发生氮质血症并伴有代谢性酸中毒等内环境紊乱。 由于 GFR下降程度不严重,肾小管部分功能还存在,但有尿浓缩功能障碍,所以尿量较多,尿钠含量较低,尿相对密度也较低。尿沉渣检查时细胞和管型较少。总体来看,非少尿型ARI肾功能障碍比少尿型ARI为轻,病程相对较短,严重并发生症少,预后较好。
少尿型和非少尿型可以相互转化:少尿型ARI经利尿、脱水治疗有可能转化为非少尿型ARI,表示病情好转,预后较好。非少尿型ARI可因治疗不及时或措施不当而转化为少尿型ARI,表示病情恶化,预后较差。

第二节 慢性肾功能不全
慢性肾功能不全(chronic renal insufficiency,CRI):任何疾病,如能使肾单位发生进行性破坏,则在数月、数年或更长时间,残存的肾单位不能充分排出代谢废物和维持内环境恒定,因而体内逐渐出现代谢废物的潴留和水、电解质与酸碱平衡紊乱以及肾内分泌功能障碍,此种情况称CRI。
一、病因
(一)肾疾患: 慢性肾小球肾炎最常见。
(二)肾血管疾患:
(三)尿路慢性梗阻:
二、发病过程及其机制
慢性肾功能不全的病程是进行性加重的,可分代偿期和非代偿期。
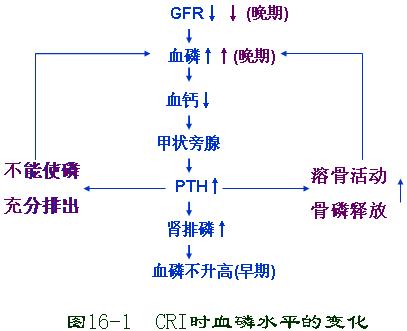
慢性肾功能不全的发病机制:
1、健存的肾单位日益减少(完整肾单位学说)健存肾单位的多少,是决定慢性肾功能不全的重要因素。
2、矫枉失衡学说。
3、肾小球过度滤过学说。
4、肾小管—肾间质损害学说。
三、对机体的影响
◆(一)泌尿功能障碍【重点】
1、尿量的变化 CRI早期出现夜尿、多尿、低渗尿。晚期可出现少尿 ,等渗尿。
(1)夜尿:正常人每日尿量约为1500ml,白天尿量约占总尿量的2/3,夜间尿量只占1/3。
夜尿(nocturia):慢性肾功能不全患者,早期即有夜间排尿增多的症状,夜间尿量和白天相近,甚至超过白天尿量,这种情况称之为夜尿。
※(2)多尿 (polyuria):每24h尿量超过2000ml时称为多尿 【重点】。是CRI早期常见的变化。
◆《多尿发生的机制》【重点】:
①CRI时,由于多数肾单位遭到破坏,流经残留的肥大肾小球的血量呈代偿性增加,因而此时滤过的原尿量超过正常
②原尿中溶质多。流速快,通过肾小管时未能及时重吸收。
③在慢性肾盂肾炎时,由于髓袢发生病变髓质间质不能形成高渗环境,使尿液不能浓缩。
(3)少尿(oliguria):在CRI晚期,当肾单位极度减少,尽管残存的有功能的每个肾单位生成尿液虽多,但生成的原尿总量还是过少,使每日终尿量少于400ml,出现少尿。
2、尿渗透压的变化
正常人尿相对密度为1、002~1、035,尿渗透压为360~1450mmol/L。
※低渗尿(hyposthenuria)【重点】:在CRI早期,肾浓缩功能减退而稀释功能正常,因而出现低相对密度尿,当尿相对密度最高只能达到1.020时,称为低渗尿。
※等渗尿(isosthenuria)【重点】:随着病情发展,到CRI晚期,肾浓缩功能和稀释功能均丧失,尿相对密度固定在1.008~1.012,尿渗透压为266~300mmol/L,称之为等渗尿。
3、尿液成分的改变
(1)蛋白尿:很多肾病疾患可使肾小球滤过膜通透性增强,致使肾小球滤出蛋白增多;或肾小管上皮细胞受损,使滤过的蛋白重吸收减少,或两者兼有之,均可出现蛋白尿(pyoteinuria)。
(2)血尿和浓尿
尿中混有红细胞时称为血尿。尿沉渣中含有大量变性白细胞时,称为浓尿。
(二)体液内环境的改变
※ 1、氮质血症(azotemia)【重点】:肾功能不全时,由于GFR下降,含氮的代谢终产物如尿素、肌肝、尿酸等在体内蓄积,因而血中非蛋白氮(NPN)的含量增加,称之为氮质血症。
2、酸中毒
★《CRI程度不同,引起酸碱平衡紊乱的类型也不同》【重点】
(1)AG正常型高血氯性代谢性酸中毒
在CRI早期,如果GFR>正常人25%,则HPO42-、SO42-等阴离子尚不至发生潴留,这时产生酸中毒主要是肾小管上皮细胞氨生成障碍使H+分泌减少,H+在体内潴留,血浆HCO3-因缓冲H+而减少。由于肾小管泌H+减少,H+-Na+交换减少,Na+随尿排出增多也伴有水排出增多,因而细胞外液容量减少,从而激活RAS,继发性醛固酮分泌增多。醛固酮可促进肾,结肠对NaCl的重吸收,因而血氯增高,而醛固酮的排H+作用因肾小管上皮细胞氨的生成障碍受限,结果发生AG正常型高血氯性代谢性酸中毒。
(2)AG增大型正常血氯性代谢性酸中毒
在严重CRI的病人,当GFR降至正常人20%以下时,血浆中的固定酸代谢产物不能由尿中排泄,特别是不能排泄在体内蓄积的磷酸和硫酸中的H+,此时血浆HCO3-浓度下降,由于HPO42-,SO42-在体内蓄积,使UA增多,AG增高,而血氯浓度无明显变化,发生AG增大型正常血氯性代谢性酸中毒。 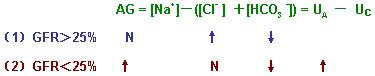
注:(1)AG正常型高血氯性代谢性酸中毒的变化
(2)AG增高型正常血氯性代谢酸中毒的变化
2、 电解质紊乱
(1)钠代谢障碍 慢性肾功能不全的肾脏都有不同程度的丢钠,尿钠含量很高,称为“失盐性肾”。失钠引起细胞外液量减少,因而进一步降低GFR,加重尿毒症。
慢性肾功能不全失钠的原因(机制):
1、渗透性利尿:CRI伴有氮质血症,通过残存肾单位排出的溶质增多,影响近曲小管对水的重吸收,同时迫使大量的钠随尿排出。此外残存肾单位的尿流速加快,也妨碍肾小管的重吸收。
3、 甲基胍蓄积:在CRI时,体内甲基胍的蓄积,可抑制肾小管对钠的重吸收。
(2)钾代谢障碍
① 血钾正常 ② 高钾血症 ③ 低钾血症
★(3)钙磷代谢障碍【重点】
1)高血磷
在CRI早期,GFR下降时血磷暂时上升,但由于钙磷乘积为一常数,使血钙降低,血中游离钙减少,刺激甲状胖腺分泌PTH,后者抑制肾小管对磷的重吸收,使磷排出增多,血磷不升高。CRI晚期,由于GFR极度下降,继发性PTH分泌增多已不能使磷充分排出,血磷升高。PTH的增多又加强溶骨活动,使骨磷释放增多,从而形成恶性循环,导致血磷不断升高。所以CRI早期血磷不升高,CRI晚期血磷升高。
2)低血钙 低血钙的机制:
①血磷升高:血浆[Ca]×[P]为一常数,血磷升高,血钙就降低([Ca]×[P]=35~40)。同时在血磷升高时,磷从肠道排出增多,在肠内与食物中的钙结合成难溶解磷酸钙排出,妨碍钙的吸收。
②维生素D代谢障碍:由于肾功能减退,肾脏羟化25-(OH)D3的功能减退,使1-25(OH)2D3合成不足从而影响肠道对钙的吸收。
③血磷升高刺激甲状旁腺C细胞分泌降钙素,抑制肠道 对钙的吸收。
④体内某些毒性物质的滞留可使小肠粘膜受损而使钙的吸收减少。
(三)其它病理生理变化
1、肾性高血压(renalhypertension)
◆《肾性高血压的类型、发病机制及其鉴别》【重点】
(1)RAS的活动增强:在某些肾疾病患者,由于肾相对缺血,激活了RAS,使血管收缩,外周阻力增加,引起高血压,称为肾素依赖性高血压(RDH)。对RDH限钠、应用利尿剂无效,可使用血管紧张素Ⅰ受体阻断剂(ATIRA):如氯沙坦(losartan)等。或血管紧张素转化酶抑制剂(ACEI):如卡托普利(captopril)等拮抗RAS活性,可以降低血压。
(2)钠水潴留:CRI时,由于肾排钠,排水功能降低, 钠水在体内潴留,血容量增加和心输出量增大,产生高血压,称为钠依赖性高血压(SDH)。对SDH限制钠盐的摄入并应用利尿剂可以降低血压。
依此与RDH进行鉴别。
(3)肾分泌的抗血压物质PGE2、PGA2减少。
2、 肾性贫血
◆ 肾性贫血的发病机制【重点】
(1)促红素生成减少:由于肾实质破坏,促红素产生减少,从而使骨髓干细胞形成红细胞受抑制,红细胞生成减少。
(2)血液中的毒性物质作用:如甲基胍对红细胞生成具有抑制作用,或者引起溶血和出血,因而加重贫血。
(3)红细胞破坏速度加快:在严重肾功能不全时,红细胞膜上Na+-K+-ATP酶受到抑制,钠泵能量供应不足,导致钠不能排出,使红细胞处于高渗状态,红细胞脆性增加,易溶血。此外,肾血管内常有纤维蛋白沉着,妨碍红细胞在血管内流动,致使红细胞易于受到机械性损伤而破裂。
(4)铁的再利用障碍:严重肾功能不全患者,虽然单核吞噬细胞系统内铁储量正常,由于铁从MPS释放受阻,血清铁浓度和铁结合力降低。
(5)出血:肾功能不全患者常有出血倾向与出血,因而加重贫血。
3、出血倾向 出血是血小板质的变化而不是数量减少所造成的。尿
毒症病人血浆中胍基琥珀酸含量增高,抑制PF3的正常释放,可能是CRI出血倾血向的主要原因。
4、肾性骨营养不良
※肾性骨营养不良(renai osteodystrophy)【重点】:是指慢性肾功能不全时,见于幼儿的肾性佝偻病,成人的骨软化,骨质疏松和骨硬化。
◆《肾性骨营养不良的发病机制》【重点】:
(1)钙磷代谢障碍和继发性甲状腺功能亢进:CRI患者由于高血磷导致血钙水平下降,刺激甲状旁腺功能亢进,分泌大量PTH,致使骨质疏松和骨硬化。
(2)维生素D代谢障碍: 在CRI患者,由于1.25-(OH)2D3合成减少,致使肠对钙磷吸收发生障碍,同时肾小管对钙磷吸收减少,致使大量磷酸盐从尿中排出,血磷降低。血磷降低影响骨和软骨基质的钙化。
(3)酸中毒:① 由于体液中H+的浓度持续升高,于是动员骨盐来缓冲,促进骨盐溶解。② 酸中毒干扰1.25-(OH)2D3的合成,抑制肠对钙磷的吸收,致使血液中钙与磷水平下降,促进肾性佝偻病或骨软化症的发生。 第三节 尿毒症
※尿毒症(uremia)【重点】:急性和慢性肾功能不全发展到最严重的阶段,代谢终末产物和内源性毒性物质在体内潴留,水、电解质和酸碱平衡发生紊乱以及某些内分泌功能失调,从而引起一系列自体中毒症状,称为尿毒症。
一、功能和代谢的变化(略)
二、发病机制
尿毒症的症状和尿毒症的毒素有关。尿毒症的毒素:
(一)甲状旁腺激素(PTH):PTH是引起尿毒症的主要毒素,能引起尿毒症的大部分症状和体症:
(二)胍类化合物:胍类化合物是体内精氨酸的代谢产物。主要是甲基胍和胍基琥珀酸。
(三)尿素:尿素的毒素作用与其代谢产物—氰酸盐有关。
(四)胺类:包括脂肪族胺、芳香族胺和多胺。
(五)中分子毒性物质:中分子毒性物质包括正常代谢产物,细胞代谢紊乱产生的多肽,细菌或细胞碎裂产物。
(六)其他:肌酐可引起溶血、嗜睡。尿酸在心包炎的发病中可引起一定作用。酚类可能是导致出血倾向的原因。 |
