第五章 缺氧
※缺氧(hypoxia):当组织得不到充足的氧,或不能利用氧时,组织的代谢、功能,甚而形态结构都可能发生异常变化,这一病理过程称为缺氧。【重点】
常用的血氧指标
1、氧分压(partial pressure of oxyge、po2):为溶解于血液的氧所产生的张力。
正常: PaO2 13.3KPa,取决于吸入气体的氧分压和肺的呼吸功能。PvO2 5.33KPa,取决于内呼吸功能状 态。
2、氧容量(oxygen binding capacity、CO2max):为100mI血液中血红蛋白为氧充分饱和时的最大带氧量。它反映血液携氧能力,取决于血液中Hb的质和量。正常:1.34mI/g×15 g/dl = 20mI/ dI
3、氧含量(oxygen content、CO2 ):为100mI血液实际的带氧量,包括物理溶解和Hb实际结合的氧量,它反映血液实际供氧水平,取决于氧分压和氧容量。正常:CaO2 19mI/dI,CvO2 14mI/dI。
4、氧饱和度(oxygen saturation、SO2):是Hb与氧结合达到饱和的程度。

正常:SaO2 95% , SvO2 70%。
SO2主要取决于PO2,PO2与SO2的关系可用氧合血红蛋白解离曲线表示。
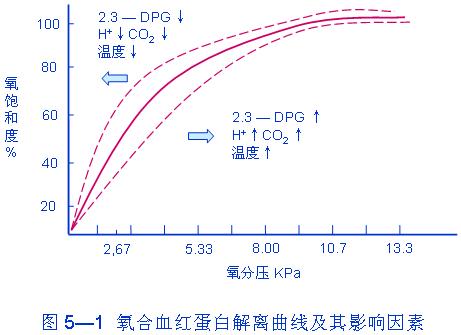
5、动―静脉血氧差(CaO2―CvO2):为动脉血氧含量减去静脉血氧含量的差值。表示组织对氧的消耗量它取决于动脉血氧含量和组织摄取氧的能力。
正常:CaO2―CvO2=19ml/dl -14 ml/dl=5 ml/dl
第一节 缺氧的类型、原因和发病机制

一、低张性缺氧
※低张性缺氧(hypotonic hypoxia):是指由于动脉血氧分压降低,使动脉血氧含量减少,以致组织供氧不足所引起的缺氧。【重点】
(一)原因
凡能使PaO2降低的原因,都能引起低张性缺氧
1、吸入气氧分压降低:又称大气性缺氧
2、外呼吸功能障碍:又称呼吸性缺氧
3、静脉血分流入动脉
★(二)血氧变化的特点与组织缺氧的机制【重点】
1、低张性缺氧时,动脉血氧分压,氧含量、氧饱和度均降低,血氧容量正常。
2. 由于氧分压在8KPa以上时氧离曲线近似水平线,在8KPa以下曲线斜率较大,所以PaO2降至8KPa以下才会SaO2及CaO2显著减少,才可能引起组织缺氧。
3.血液中的氧弥散入细胞线粒体的速度决于血液与线粒体部位的氧和分压差。若PaO2与CaO2过低使氧弥散速度减慢,可引起细胞缺氧。
4、低张性缺氧时CaO2降低,由同量血液弥散给组织利用的氧量减少,因此动―静脉氧差一般是减小的。如慢性缺氧使组织利用氧的能力代偿性增强,则动―静脉氧差也可正常。
※5、发绀(cyanosis):毛细血管中脱氧血红蛋白平均浓度增加到5g/dI以上可使皮肤与粘膜呈青紫色,称为发绀(紫绀)。【重点】
低张性缺氧时,动脉血与静脉血的氧合血红蛋白浓度均降低,毛细血管中脱氧血红蛋白浓度增加,容易引起发绀。发绀是缺氧的表现,但缺氧的人不一定都发绀。
二、血液性缺氧
※血液性缺氧(hemic hypoxia):是由于血红蛋白数量减少或性质改变,以致血氧含量降低或血红蛋白结合的氧不易释出所引起的组织缺氧。因为这种缺氧动脉血氧分压正常,又称等张性低氧血症。【重点】
(一)原因
1、贫血:又称贫血性缺氧
2、一氧化碳中毒
★《一氧化碳中毒引起缺氧的机制》【重点】
① CO与血红蛋白结合形成碳氧血红蛋白(CO+Hb→HbCO),从而失去了运氧功能,组织发生缺氧。
②抑制红细胞内的糖酵解作用,使2,3-DPG生成减少,引起氧离曲线左移,使HbO2中的氧不易释放,从而加重组织缺氧。
3、高铁血红蛋白血症:较常见于亚硝酸盐中毒
※ ★《亚硝酸盐中毒引起缺氧的机制》【重点】
亚硝酸盐是一种氧化剂,可以把血红蛋白中的二价铁离子氧化成三价铁离子,形成高铁血红蛋白(methemoglobin, HbFe3+OH)。高铁血红蛋白中的三价铁离子与羟基牢固结合而失去了携氧能力,引起缺氧。同时在高铁血红蛋白中因三价铁离子使剩余的二价铁离子与O2的亲合力增强,氧解离曲线左移,加重组织缺氧。
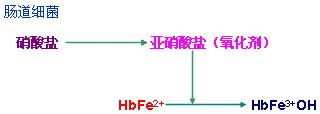
食用大量含硝酸盐的腌菜后,经肠道细菌将硝酸盐还原为亚硝酸盐,后者吸收导致高铁血红蛋白血症,称为肠源性紫绀。
4、血红蛋白与氧的亲和力异常: ① 输入大量库存血液;② 输入大量碱性液体;③ 某些血红蛋白病。
★(二)血氧变化特点与组织缺氧的机制【重点】
1、血液性缺氧时,由于外呼吸功能正常,动脉血氧分压及血氧饱和度正常,但因Hb的数量减少或性质改变,使血氧容量降低,血氧含量减少。由于动脉血氧含量低,而组织摄取氧的能力正常,动―静脉血氧含量差减少。
Hb与O2亲合力增强引起的血液性缺氧较特殊,其动脉血氧容量和血氧含量可正常,甚至有的还高于正常。由于Hb和O2的亲和力较大,结合氧不易释出,其动―静脉血氧差也小于正常。〖见表5-1〗
2、血液性缺氧的病人可无发绀
(1)严重贫血的病人,Hb数量减少,面色苍白,毛细血管中脱氧血红蛋白达不到5g/dI ,不会出现发绀。
(2)一氧化碳中毒者,因HbCO呈樱桃红色,使皮肤、粘膜呈樱桃红色。但重度中毒,严重缺氧时,由于皮肤血管收缩,皮肤粘膜呈苍白色。
(3)高铁血红蛋白呈咖啡色或青石板色,使患者皮肤呈咖啡色或类似发绀的颜色,但不是发绀。
(4)单纯由Hb与O2亲合力增高引起的缺氧,毛细血管中脱氧血红蛋白少于正常,因此不能出现发绀。
三、循环性缺氧
※循环性缺氧(circulatory hypoxia):由于组织血流量减少使组织供氧量减少所引起的缺氧。或称低动力性缺氧。【重点】
循环性缺氧可分为缺血性缺氧、淤血性缺氧。
(一)原因
不论是全身的血液循环障碍,见于休克和心力衰竭。还是局部血液循环障碍,见于栓塞,脉管炎等血管病变,都可使组织血流量减少,引起循环性缺氧。
★(二) 血氧变化的特点与组织缺氧的机制【重点】
1、单纯性循环性缺氧时,动脉血氧分压,血氧饱和度,血氧容量,血氧含量是正常的。
由于血流缓慢,血液流经毛细血管的时间延长,从单位容积血液弥散给组织的氧量较多,静脉血氧含量降低,使动-静脉血氧差增大。但单位时间内流经毛细血管的血量减少,弥散到组织细胞的氧量减少,导致缺氧。〖见表5-1〗
2、由于静脉血的氧含量和氧分压较低,毛细血管中脱氧血红蛋白可超过5g/dI,因而引起发绀。
四、组织性缺氧
※组织性缺氧(dysoxidative hypoxia):由组织细胞利用氧障碍所引起的缺氧。【重点】
(一)原因
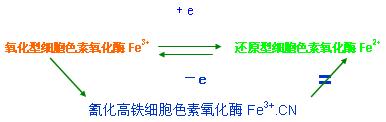
1、组织中毒 如氰化物、硫化物、砷化物、甲醇、鱼藤酮和某些药物(巴比妥,抗霉菌素,苯已双胍等)。
◆《氢化物中毒引起缺氧的机制》【重点】
各种氰化物可由消化道、呼吸道或皮肤进入体内,迅速与氧化型细胞色素氧化酶的三价铁结合为氰化高铁细胞色素氧化酶,使之不能还原成还原型细胞色素氧化酶,以致呼吸链中断,组织不能利用氧。大量服用氰化物可造成“闪电式”死亡!
2、细胞损伤 大量放射线照射、细菌毒素(内毒素)损伤线粒体、
吸入高压氧、组织严重供氧不足抑制线粒体呼吸功能。
3、呼吸酶合成障碍
硫 胺 素(B1) 组成 丙酮酸脱氢酶辅酶
尼克酰胺(PP) 组成 NAD+、NADP+
核 黄 素(B2) 组成 黄素辅酶
这些维生素严重缺乏可能导致氧的利用障碍。
★(二)血氧变化的特点【重点】〖见表5-1〗
组织性缺氧时,动脉血氧分压、氧饱和度,血氧容量、血氧含量一般均正常。由于内呼吸障碍使组织不能充分利用氧,使静脉血氧含量较高,动-静脉血氧差小于正常。由于组织细胞利用氧发生障碍,毛细血管中氧合血红蛋白含量高于正常,所以组织性缺氧不会有紫绀。
●缺氧可分为上述四种类型,其中哪些类型的缺氧有紫绀,而哪些类型的缺氧无紫绀,为什么有此不同?【重点】
低张性缺氧和循环性缺氧可以引起发绀。血液性缺氧和组织性缺氧不易发绀。低张性缺氧时,动脉和静脉中氧合血红蛋白浓度均减少,全血中脱氧血红蛋白浓度增加。当毛细血管中脱氧血红蛋白浓度增加到5g/dl以上时,可使皮肤与粘膜呈青紫色,出现发绀。循环性缺氧时,因血液流经毛细血管时间延长,从单位容量血液弥散给组织的氧量增加,故静脉血氧含量明显降低。毛细血管中脱氧血红蛋白含量可超过5g/dl,所以可出现发绀。血液性缺氧无发绀,因为严重贫血的病人,Hb数量减少,面色苍白,毛细血管中脱氧血红蛋白达不到5g/dl,不会出现发绀。CO中毒,因HbCO呈樱桃红色,皮肤、粘膜呈现樱桃红色,但重度中毒,严重缺氧,由于皮肤血管收缩,皮肤、粘膜可呈苍白色。高铁血红蛋白使皮肤、粘膜呈现咖啡色或青石板色,但不是发绀。在因Hb与O2亲和力增强引起的血液性缺氧时,动脉血氧容量和氧含量可不低甚至还稍高于正常。其动-静脉血氧含量差缩小,毛细血管中脱氧血红蛋白含量不会超过5g/dl,所以不会引起发绀。组织性缺氧时因内呼吸功能障碍使组织不能充分利用氧,故静脉血氧含量和氧分压较高,故毛细血管中脱氧血红蛋白含量不会超过5g/dl,所以不会引起发绀。
●临床上所见的缺氧往往是混合型缺氧。现以失血性休克、感染性休克为例,分析感染性休克,失血性休克可能会发生几种类型的缺氧并简要说明其机制。【重点】
感染性休克:由于休克引起微循环的改变,导致全身循环功能障碍,使组织供氧量减少,可发生循环性缺氧。严重休克可使胃肠功能紊乱,胃肠粘膜屏障功能丧失,细菌内毒素入血,引起组织细胞损伤,使组织利用氧障碍,发生组织性缺氧。晚期并发休克肺时,外呼吸功能障碍引起低张性缺氧。
失血性休克:由于休克引起微循环的改变,导致全身循环功能障碍,使组织供氧量减少,可发生循环性缺氧。大量失血,引起贫血,导致血液性缺氧。严重休克可使胃肠功能紊乱,胃肠粘膜屏障功能丧失,细菌内毒素入血,引起组织细胞损伤,组织利用氧障碍,发生组织性缺氧。晚期并发休克肺时,外呼吸功能障碍引起低张性缺氧。
第二节 缺氧时机体的功能代谢变化
一、呼吸系统变化
(一)代偿性反应
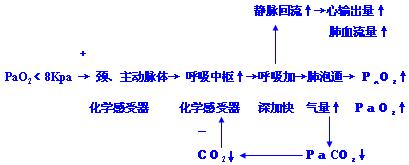
◆《低张性缺氧所引起的肺通气变化与缺氧持续的时间有关》【重点】PaO2降低(小于8kPa)可刺激颈动脉体和主动脉体化学感受器,反射性引起呼吸加深加快,从而使肺通气量立即增加。但是,在急性缺氧早期肺通气量增加较少,可能因过度通气形成的低碳酸血症和呼吸性碱中毒对呼吸中枢的抑制作用,使肺通气的增加受阻。2―3日后肺通气量显著增加,这是由于肾代偿性排出HCO3-,脑脊液中HCO3-的也逐渐通过血脑屏障进入血液,使脑组织中pH逐渐恢复正常,此时方能显示缺氧兴奋呼吸的作用。长期缺氧使肺通气量回降,可能与外周化学感受器对缺氧的敏感性降低有关。
(二)呼吸功能障碍
1、高原肺水肿:表现为呼吸困难,咳嗽,咳血性泡沫痰,肺部有湿性罗音,皮肤粘膜发绀等。机制不清。患者下到平原地区,氧疗后可全愈,不留后遗症。
2、中枢性呼吸衰竭
二、循环系统的变化
(一)代偿性反应
1、心输出量增加
(1)心率加快
(2)心肌收缩性增强
(3)静脉回流增加
2、 血流分布改变
3、肺血管收缩
◆《缺氧引起肺血管收缩的代偿意义及其机制》【重点、难点】
无论是肺泡缺氧还是混合静脉血的氧分压降低都引起肺小动脉收缩,从而导致缺氧的肺胞的血流量减少。
代偿意义:由肺泡通气量减少引起的局部肺血管收缩反应有利于维持肺泡通气与血流的适当比利,使流经这部分肺泡的血液仍能获得较充分的氧,从而可维持较高的PaO2。此外,正常情况下由于重力作用,肺尖部的肺泡通气量与血流量的比值过大,肺泡气中氧不能充分被血液运走。当缺氧引起较广泛的肺血管收缩导致肺动脉压升高时,肺上部的血流增加,肺上部的肺泡通气能得到更充分的利用。
肺血管收缩的机制:
(1)交感神经作用: 缺氧引起的交感神经兴奋可作用于肺血管的a受体引起血管收缩反应。
(2)体液因素作用:缺氧可促使肺组织内肥大细胞、肺巨噬细胞、血管内皮细胞、血细胞、乃至血管平滑肌细胞释放血管活性物质。收缩血管的有:如白三稀(leukotriene, LTs);血栓素A2(thromboxane A2, TXA2),内皮素(endothelin, ET)。舒张血管的有:如前列环素(prostacyclin, PGI2),内皮源性舒张因子(endothelium derived relaxing factor, EDRF);组胺(histamin)等。在肺血管收缩反应中,缩血管物质生成增加起介导作用,扩血管物质生成也增加,起调节作用,两者力量的对比决定肺血管收缩反应的强度。显然,缩血管物质作用占优势。
(3)对血管平滑肌直接作用:缺氧使平滑肌细胞钾通道关闭,外向性K+电流减少,膜电位下降,膜去极化,再导致电压依赖性钙通道开放,Ca2+内流增加引起肺血管收缩。
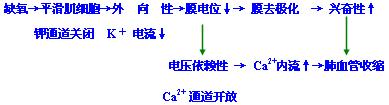
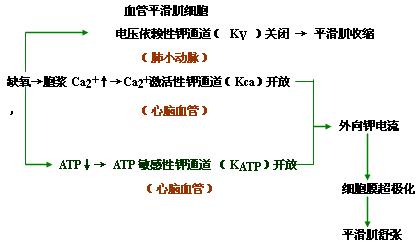
4、血管对缺氧反应的异质性不同的血管对缺氧的反应性不同,肺小动脉呈收缩反应,心脑血管呈舒张反应。
●《缺氧时心脑血管呈舒张反应(血管对缺氧反应的异质性)的表现及其机制》【重点、难点】
不同的血管对缺氧的反应不相同,除上述因素外,还与血管平滑肌细胞的钾通道分布有关。血管平滑肌细胞上有电压依赖性钾通道(Kv),Ca2+激活性钾通道(KCa)和ATP敏感性钾通道(KATP)。缺氧使Kv关闭引起平滑肌收缩;胞浆游离钙增加致KCa开放;ATP减少使KATP开放,后两者均可增加外向钾电流,引起细胞膜超极化,致平滑肌松弛和血管舒张。肺小动脉平滑肌细胞以含Kv为主的多,故对缺氧呈收缩反应;心、脑血管平滑肌细胞以含KCa和KATP为主的多,故对缺氧呈舒张反应。
5.毛细血管增生 长期缺氧可促使血管内皮生长因子(VEGF)等基因表达增加,使毛细血管增生。
(二)循环功能障碍
▲《以高原性心脏病为例说明缺氧引起循环障碍的机制》【重点】
(1) 肺动脉高压:由于缺氧时肺血管的强烈收缩,使肺循环阻力增加,形成肺动脉高压;慢性缺氧使肺小动脉长期处于收缩状态,引起肺血管中膜平滑肌肥大,血管硬化,形成持续的肺动脉高压。缺氧引起红细胞增多,使血液粘度增高也可增加肺血流阻力。肺动脉高压增加了右心射血的阻力,出现右心肥大,久之可发生右心衰竭,全心衰竭。
(2) 心肌的收缩与舒张功能降低:严重缺氧时,心肌产生能量代谢障碍,心肌舒缩功能降低;严重缺氧可引起心肌变性坏死。
(3) 心律失常:严重缺氧可引起心动过缓,期前收缩,甚至发生心室纤颤致死。
(4) 静脉回流减少:严重缺氧时,呼吸中枢的抑制使胸廓运动减弱,致静脉回流减少;全身缺氧时,体内乳酸,腺苷等代谢产物堆积,对外周血管有直接扩张作用,使外周血管床扩大,大量血液淤积在外周,回心血量减少,使心输出量减少。
三.血液系统变化
1、红细胞(RBC)增多
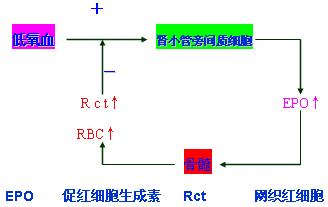
◆《慢性缺氧引起红细胞增多的机制及其代偿意义》【重点】
答:慢性缺氧可使红细胞生成增多。因为低氧血流经肾脏时能刺激肾小管旁间质细胞生成并释放促红细胞生成素(erythropoietin)。后者促进骨髓造血机能增强促进干细胞分化为原红细胞、促进其分化和成熟,加速Hb合成,及骨髓内网织红细胞和红细胞释放入血。代偿意义:红细胞增多可增加血氧容量和氧含量,从而增加组织供氧量。
2.氧离曲线右移
缺氧时红细胞 内2.3-DPG增加,导致氧离曲线右移
●(1)缺氧时红细胞生成的2、3-DPG增多的机制【难点】
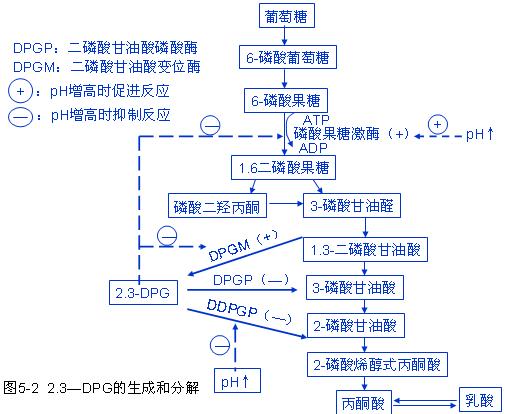
①缺氧时,氧合血红蛋白减少,而脱氧血红蛋白增多,后者中央孔穴比前者大,可结合2,3-DPG,使红细胞内游离的2,3-DPG减少,因而减少了对磷酸果糖激酶及二磷酸甘油变位酶的抑制作用,导至糖酵解过程增强,2,3-DPG生成增加。
②由于脱氧Hb稍偏碱性和代偿性肺通气过度引起呼吸性碱中毒,使pH增高,后者能激活磷酸果糖激酶使糖酵解过程增强,2,3-DPG生成增多。
③高pH还能抑制2,3-DPG磷酸酶的活性,使2,3DPG分解减少。●(2)2、3-DPG增多使氧离曲线右移的机制 【难点】
① 2、3-DPG与脱氧血红蛋白结合,使脱氧血红蛋白的空间结构稳定,不易与O2结合。
② 2、3-DPG是一种不能透出红细胞膜的有机酸,增多使红细胞内pH降低,pH值下降可通过Bohr效应使血红蛋白与氧的亲和力降低,也使曲线右移。
四、中枢神经系统的变化
急性缺氧:头痛、情绪激动、思维力、记忆力、判断力降低或丧失以及运动不协调等。
慢性缺氧:疲劳、嗜睡、注意力不集中及精神抑郁等症状。
严重缺氧:烦燥不安、惊厥、昏迷、甚而死亡。
正常:脑静脉血氧分压 ⒋53 KPa
降 至 ⒊73 KPa 精神错乱
降 至 ⒉53 KPa 意识丧失
降 至 ⒈60 KPa 危及生命
脑组织形态学变化:脑细胞肿胀变性、坏死及脑间质水肿。
五、组织细胞变化
(一)代偿性反应
1、组织细胞利用氧的能力增强
2、无氧酵解增强
3、肌红蛋白增加
4、低代谢状态
●5、细胞对缺氧的反应及其机制【难点】
(1)细胞对缺氧的反应
①颈动脉体化学感受器在缺氧时分泌神经介质,引起反射性呼吸运动增强。
②血管平滑肌细胞对缺氧发生的舒缩反应,可改变血流分布。
③缺氧时肾小管间质细胞产生促红细胞生成素,使骨髓红细胞生成增多。
④细胞缺氧时血管内皮生长因子等基因表达增强,促进血管增生。
这些细胞反应可提高机体对缺氧的适应能力,有利于整体的生存。
(2)细胞对缺氧反应的机制
细胞缺氧时,通过改变细胞的氧化还原状态,活性氧生成的减少,NADH/NAD+,NADPH/NADP+和GSH/GSSH的比例增高,使胞浆内缺氧诱导因子-1及其他转录因子被激活,与靶基因增强子或启动子结合,对基因表达起促进作用,导致蛋白质合成的改变,从而影响细胞的代谢功能,引起细胞的缺氧反应。
此外,缺氧时细胞氧化还原状态的改变也可能直接影响离子通道的开关,导致细胞膜电位及功能变化。
缺氧诱导因子-1(hypoxia inducible factor-1,HIF-1)是细胞在缺氧条件下产生的核蛋白。
HIF-1的靶基因主要包括以下几种:
①促红细胞生成素(EPO)编码基因
②血管内皮生长因子(VEGF)编码基因
③葡萄糖载体蛋白-1(GLUT-1)和糖酵解编码基因
④血红素加氧酶(HO-1)和诱导型NO合酶(INOS)编码基因
缺氧条件下,细胞核产生HIF-1或其他转录因子被激活,与靶基因结合,促进基因转录,引起一系列细胞对缺氧的反应,在促进红细胞生成,血管生长,促进糖酵解及调节血管舒缩方面具有重要的作用,对保持机体的氧稳态,具有重要代偿意义。
(二)细胞损伤
●《缺氧性细胞损伤主要的哪些变化及其变化的机制》【难点】
缺氧性细胞损伤主要表现为细胞膜、线粒体及溶酶体的变化。
(1) 细胞膜变化
由于严重缺氧使细胞膜性结构的磷脂部分代谢障碍,膜完整性受损,通透性增大,膜稳定性下降。导致细胞内外离子顺浓度差运动。
①Na+离子内流增多:Na+泵因缺氧能量不足而不能泵出细胞内过多的Na+,导致细胞水肿。血管内皮细胞肿胀可堵塞微血管,加重组织缺氧。
②K+离子外流增多:引起细胞内缺钾,而K+是蛋白质合成代谢所必需的,因此引起蛋白质包括酶蛋白合成代谢障碍,进一步影响ATP生成和离子泵功能。
③钙离子内流增多:细胞外Ca2+浓度比细胞内高1000倍,当严重缺氧时,细胞膜通透性升高,Ca2+离子内流增加。ATP减少,钙泵失灵影响Ca2+的泵出和被摄取。胞浆中Ca2+增多可抑制线粒体呼吸功能,可激活磷脂酶,使膜磷脂分解,引起溶酶体酶释出;还可激活一种钙依赖性蛋白水解酶,使黄嘌呤脱氢酶转为黄嘌呤氧化酶,增加自由基的形成,加重细胞损伤。
(2) 线粒体变化
轻度缺氧或缺氧早期线粒体的呼吸功能代偿性增强。严重缺氧时,首先影响线粒体外的氧利用。使神经介质的生成和生物转化过程降低。当线粒体部位氧分压降到临界点(0.1kPa(1mmHg)时,线粒体呼吸功能下降,ATP生成减少。严重时,线粒体本身出现肿胀、嵴断裂、外膜破裂和基质外溢等变化。
(3) 溶酶体变化
主要表现为溶酶体膜磷脂分解,通透性升高。最后肿胀、破裂和大量溶酶体酶释出,导致细胞及周围组织溶解、坏死。溶酶体变化的主要原因是糖酵解增强,乳酸生成增多;及脂肪氧化不全,酮体等生成增多。导致酸中毒。pH降低使磷脂酶活性增高,破坏溶酶体膜。
|
